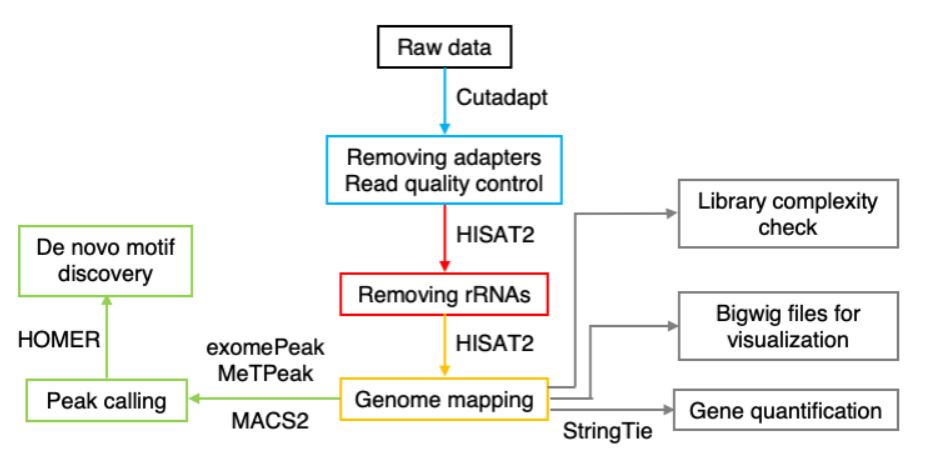
MeRIP-Seq数据分析
MeRIP-Seq数据分析
(一)数据下载
1
2
3
4
5
6
7
8
9
10
11
12
|
nohup preftch SRR9646136 &
nohup preftch SRR9646140 &
nohup preftch SRR9646137 &
fastq-dump /path/to/xxx.sra
ls *.sra|while read id; do nohup fastq-dump --split-3 $id; done
|
(二) 数据比对
step 1、质量控制
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
fastqc -t 6 *fastq -o ./qc
cd ./qc
multiqc *.zip -o ./
cat fq.txt|while read id
do
arr=(${id})
fq1=${arr[0]}
fq2=${arr[1]}
nohup trim_galore -q 20 --phred33 --length 20 -e 0.1 --stringency 3 --paired -o ../clean $fq1 $fq2 &
done
|
step 2、HISAT2比对
①、下载hisat2索引文件(含转录组信息)
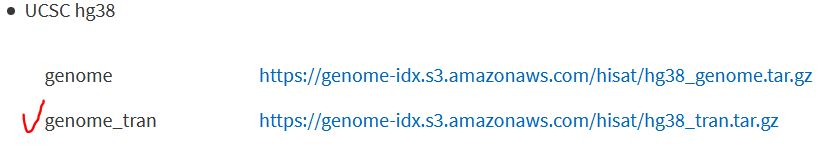
首先去HISAT2官网下载比对所需要的索引文件。注意啦,因为我们是MeRIP-seq数据的比对,构建的索引需要把转录组信息加进去,所以我们这里下载UCSC的hg38:genome_tran。注意了,这里选择UCSC的参考基因组,因为后面peak注释用到的工具——HOMER或者R包ChIPseeker内置的都是UCSC的参考基因组。如果你非要用到其他数据库的参考基因组,如NCBI的GRCh38,你就会在peaks注释阶段遇到染色体号相关的报错。当然,这个报错动动脑子也是可以解决的。
你也可以自己构建带有转录组信息的索引文件,HISAT2官网同样给出了详细的教程。但是,这个操作至少需要160G的内存😂,还是算了吧~~~没必要重复造轮子
②、下载rRNA的fastq文件,构建rRNA文件的索引
参考资料:https://cloud.tencent.com/developer/article/1805475
1
2
3
4
5
6
7
8
9
10
11
|
wget https://cloud.biohpc.swmed.edu/index.php/s/oTtGWbWjaxsQ2Ho/download
unzip download
echo 'export PATH=/home/gongyuqi/biosoft/HISAT2/hisat2-2.2.1:$PATH' >> ~/.bashrc
source ~/.bashrc
cd /home/gongyuqi/ref/rRNA
hisat2-build -p 4 rRNA.fasta ./index_hisat2/rRNA
|
③、去除rRNAs
1
2
3
4
5
6
7
8
9
10
11
12
|
for i in {36,40,37}
do
nohup hisat2 -x /home/gongyuqi/ref/rRNA/index_hisat2/rRNA \
-1 SRR96461${i}_1_val_1.fq \
-2 SRR96461${i}_2_val_2.fq \
--un-conc ../rmr_rRNA/SRR96461${i}_rmr_%.fq \
-p 16 -S ../rmr_rRNA/SRR96461${i}.sam &
done
|
④、HISAT2比对含有转录组信息的参考基因组
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
ls *_1.fq> rmr_fq1.txt
ls *_2.fq> rmr_fq2.txt
paste rmr_fq1.txt rmr_fq2.txt > rmr_fq.txt
ref=/home/gongyuqi/ref/hg38/index/hisat2_UCSC/hg38_tran/genome_tran
cat rmr_fq.txt|while read id
do
arr=(${id})
fq1=${arr[0]}
fq2=${arr[1]}
sample=${fq1%%_*}.sort.bam
nohup hisat2 -p 8 -x $ref -1 $fq1 -2 $fq2 | samtools sort -@ 8 -o ../align/$sample - &
done
nohup hisat2 -p 8 -x $ref -1 SRR9646139_rmr_1.fq -2 SRR9646139_rmr_2.fq | samtools sort -@ 8 -o ../align/SRR9646139.sort.bam - &
ls *.bam | while read id; do nohup samtools flagstat $id & done
ls *.bam | while read id; do nohup samtools index $id & done
ls *.bam | while read id; do nohup bamCoverage -b $id -o ${id%%_*}.bw & done
|
(三)MACS2 call peaks
MACS2官网参考资料
1
2
3
4
5
6
7
|
macs2 callpeak -t SRR9646136.sort.bam -c SRR9646137.sort.bam -f BAM -g hs -n siControl_m6A_IgG_replicate1 -B -q 0.01 --outdir siControl_m6A_IgG
macs2 callpeak -t SRR9646140.sort.bam -c SRR9646137.sort.bam -f BAM -g hs -n siControl_m6A_IgG_replicate2 -B -q 0.01 --outdir siControl_m6A_IgG
macs2 callpeak -t SRR9646138.sort.bam -c SRR9646139.sort.bam -f BAM -g hs -n siFTO_m6A_IgG -B -q 0.01 --outdir ../peaks
|
(四)peaks注释
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
| library(org.Hs.eg.db)
library(ChIPseeker)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
files <- list(replicate1="siControl_m6A_IgG_replicate1_summits.bed",
replicate2="siControl_m6A_IgG_replicate2_summits.bed")
peakAnno_replicate1 <- annotatePeak(files[[1]], TxDb=txdb, annoDb="org.Hs.eg.db")
peakAnno_replicate2 <- annotatePeak(files[[2]], TxDb=txdb, annoDb="org.Hs.eg.db")
siFTO_siControl_peak<-peakAnno_siFTO_siControl_m6A@anno@elementMetadata@listData
siFTO_siControl_peak<-as.data.frame(siFTO_siControl_peak)
replicate1_peak<-as.data.frame(peakAnno_replicate1@anno@elementMetadata@listData)
replicate1_peak_SYMBOL<-peakAnno_replicate1@anno@elementMetadata@listData$SYMBOL
replicate1_peak_SYMBOL<-unique(na.omit(replicate1_peak_SYMBOL))
replicate2_peak<-as.data.frame(peakAnno_replicate2@anno@elementMetadata@listData)
replicate2_peak_SYMBOL<-peakAnno_replicate2@anno@elementMetadata@listData$SYMBOL
replicate2_peak_SYMBOL<-unique(na.omit(replicate2_peak_SYMBOL))
common_peaks<-intersect(replicate1_peak_SYMBOL,replicate2_peak_SYMBOL)
table("CCND1"%in%common_peaks)
|
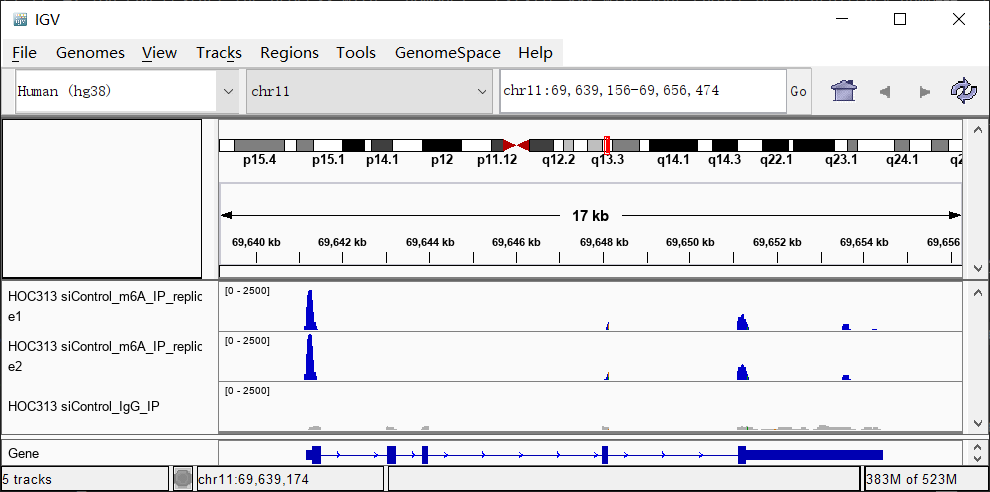
将bam文件导入IGV,可视化CCND1上m6A的情况