体验Seraut包处理GSE111229的scRNA-seq数据
1 2 rm( list = ls( ) ) Sys.setenv( R_MAX_NUM_DLLS= 999 )
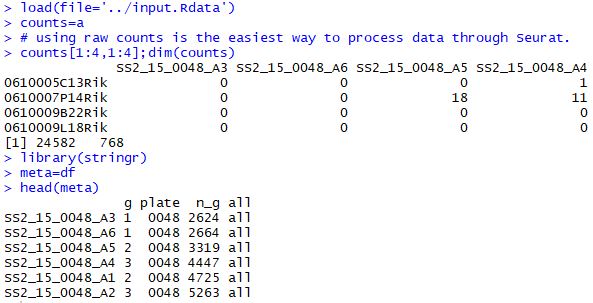
载入数据 1 2 3 4 5 6 7 8 9 10 load( file= '../input.Rdata' ) counts= a counts[ 1 : 4 , 1 : 4 ] ;dim ( counts) library( stringr) meta= df head( meta) gs= read.table( 'top18-genes-in-4-subgroup.txt' ) [ , 1 ] gs
1 2 3 4 5 fivenum( apply( counts, 1 , function ( x) sum ( x> 0 ) ) ) boxplot( apply( counts, 1 , function ( x) sum ( x> 0 ) ) ) fivenum( apply( counts, 2 , function ( x) sum ( x> 0 ) ) ) hist( apply( counts, 2 , function ( x) sum ( x> 0 ) ) )
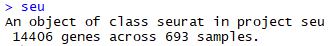
构建Seraute对象 1 2 3 4 5 6 7 8 9 library( Seurat) seu <- CreateSeuratObject( raw.data = counts, meta.data = meta, min.cells = 5 , min.genes = 2000 , project = "seu" ) seu
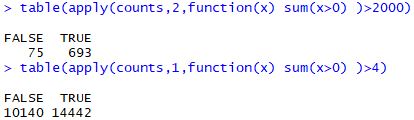
1 2 3 table( apply( counts, 2 , function ( x) sum ( x> 0 ) ) > 2000 ) table( apply( counts, 1 , function ( x) sum ( x> 0 ) ) > 4 )
可以看到,两种质控方法得到的数据非常接近
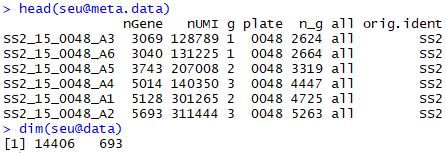
查看对象的基本情况
1 2 head( seu@ meta.data) dim ( seu@ data)
可视化基本信息
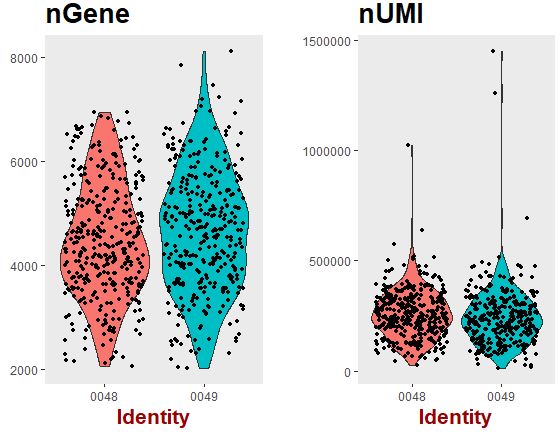
1 2 3 4 VlnPlot( object = seu, features.plot = c ( "nGene" , "nUMI" ) , group.by = 'plate' , nCol = 2 )
可以看出:不管是各个样本检测到的基因数量还是文库大小,都没有批次效应。
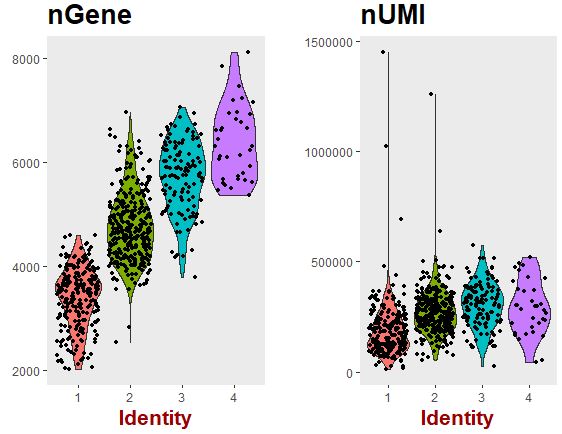
1 2 3 4 VlnPlot( object = seu, features.plot = c ( "nGene" , "nUMI" ) , group.by = 'g' , nCol = 2 )
可以看出:层次聚类得到的分组主要是因为样本间检测的基因数量的差异造成的。
可以给seu对象增加一个属性,供QC使用
1 2 3 4 5 6 7 8 9 10 ercc.genes <- grep( pattern = "^ERCC-" , x = rownames( x = seu@ raw.data) , value = TRUE ) percent.ercc <- Matrix:: colSums( seu@ raw.data[ ercc.genes, ] ) / Matrix:: colSums( seu@ raw.data) seu <- AddMetaData( object = seu, metadata = percent.ercc, col.name = "percent.ercc" ) VlnPlot( object = seu, features.plot = c ( "nGene" , "nUMI" , "percent.ercc" ) , group.by = 'g' , nCol = 3 )
可以看出:细胞能检测到的基因数量与其含有的ERCC序列反相关。当组间的文库大小差异可忽略时,外源RNA多,计算后的内源RNA的比例自然是会下降的。
进一步可视化基本信息
1 2 3 4 5 6 VlnPlot( object = seu, features.plot = c ( "nGene" , "Gapdh" , "Bmp3" ) , group.by = 'plate' , nCol = 3 )
再次证明没有批次效应
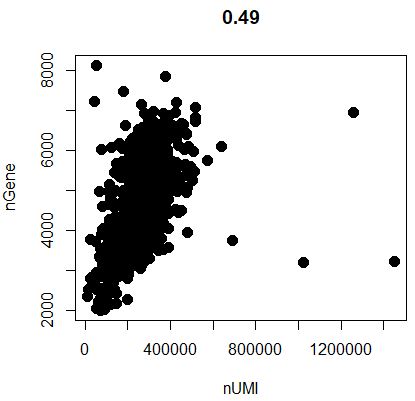
1 GenePlot( object = seu, gene1 = "nUMI" , gene2 = "nGene" )
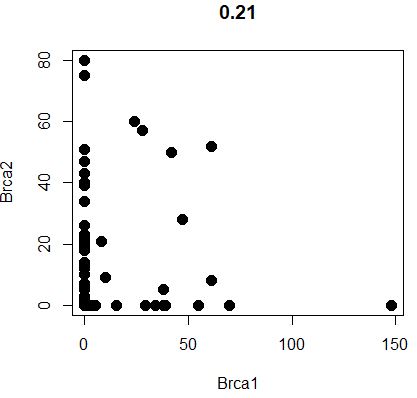
1 GenePlot( object = seu, gene1 = "Brca1" , gene2 = "Brca2" )
1 2 3 CellPlot( seu, seu@ cell.names[ 3 ] , seu@ cell.names[ 4 ] , do.ident = FALSE )
表达矩阵标准化 1 2 3 4 seu <- NormalizeData( object = seu, normalization.method = "LogNormalize" , scale.factor = 10000 ) seu@ data[ 1 : 4 , 1 : 4 ]
挑选变化的基因 1 2 3 4 5 6 7 seu <- FindVariableGenes( object = seu, mean.function = ExpMean, dispersion.function = LogVMR ) length ( seu@ var.genes)
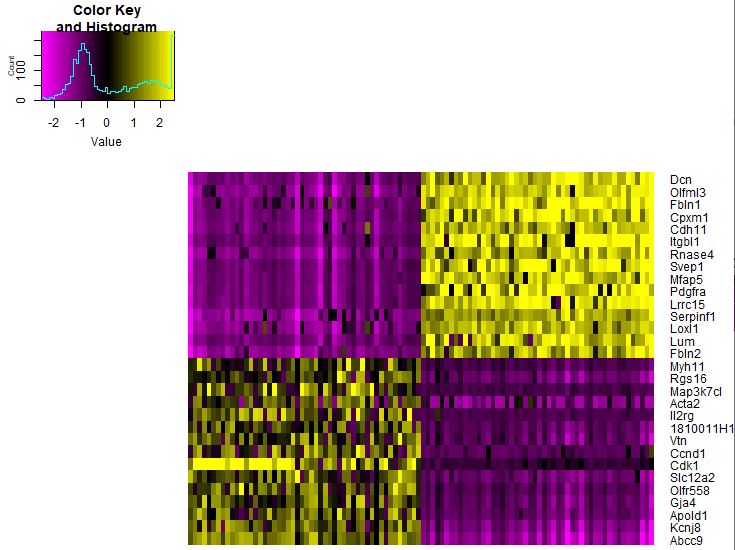
去除一些技术误差,比如 nUMI或者ERCC 1 2 3 4 5 6 head( seu@ meta.data) seu <- ScaleData( object = seu, vars.to.regress = c ( "nUMI" , 'nGene' , "percent.ercc" ) ) seu@ scale.data[ 1 : 4 , 1 : 4 ] pheatmap( as.matrix( seu@ scale.data[ gs, ] ) )
普通PCA降维 用前面挑选的有变化的基因进行降维
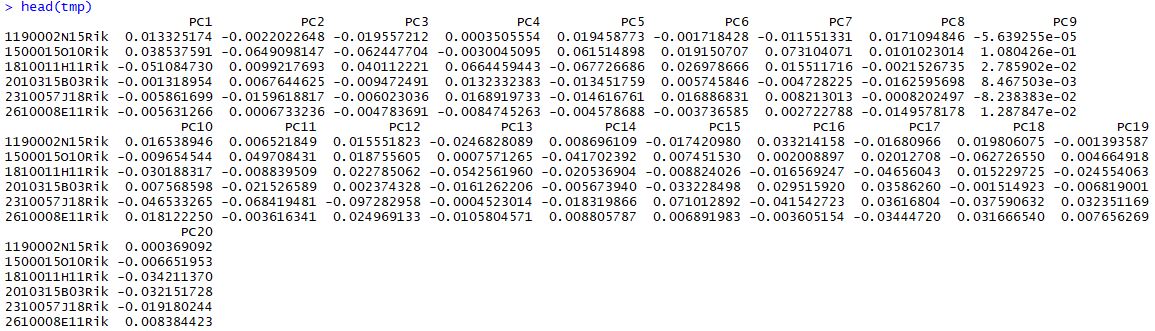
1 2 3 4 5 seu <- RunPCA( object = seu, pc.genes = seu@ var.genes, do.print = TRUE , pcs.print = 1 : 5 , genes.print = 5 ) tmp <- seu@ dr$ pca@ gene.loadings head( tmp)
1 VizPCA( seu, pcs.use = 1 : 2 )
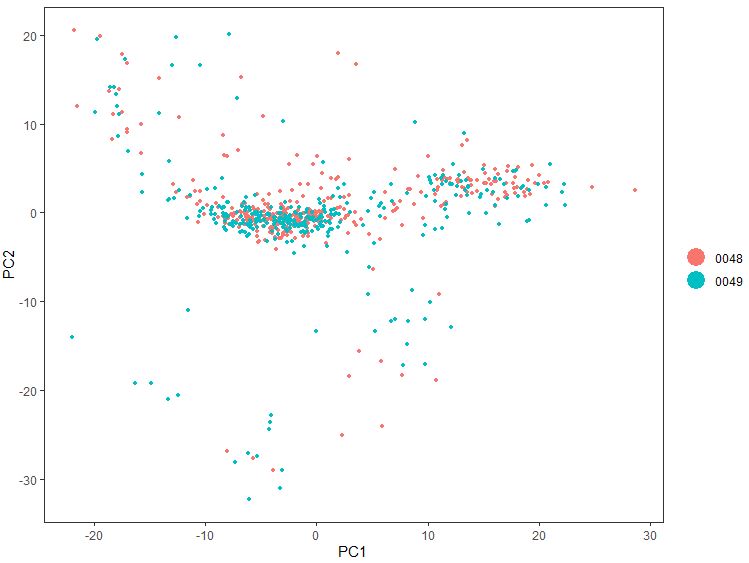
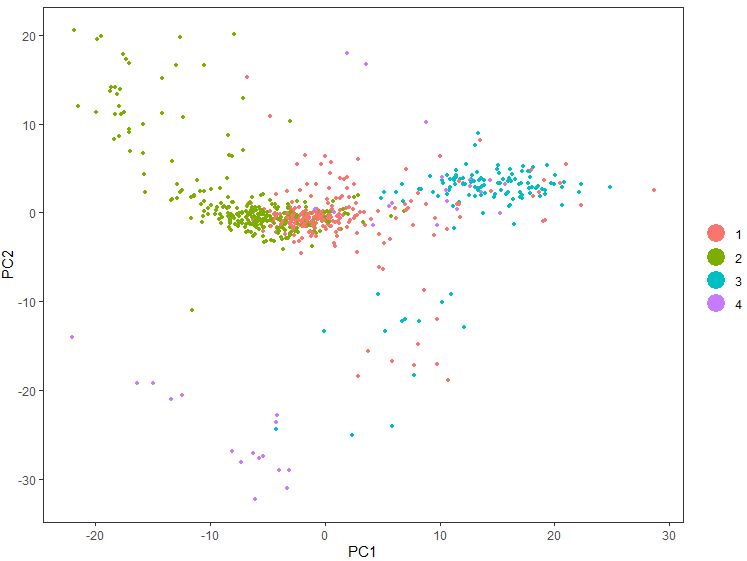
1 PCAPlot( seu, dim.1 = 1 , dim.2 = 2 , group.by = 'plate' )
1 PCAPlot( seu, dim.1 = 1 , dim.2 = 2 , group.by = 'g' )
根据不同组成份进行热图的展示
1 2 3 4 5 seu <- ProjectPCA( seu, do.print = FALSE ) PCHeatmap( object = seu, pc.use = 1 , cells.use = 100 , do.balanced = TRUE , label.columns = FALSE )
根据参数来调整最后的分组
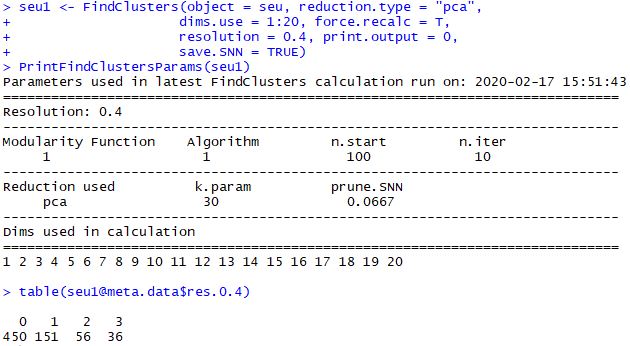
1 2 3 4 5 6 7 8 9 seu1 <- FindClusters( object = seu, reduction.type = "pca" , dims.use = 1 : 20 , force.recalc = T , resolution = 0.4 , print.output = 0 , save.SNN = TRUE ) PrintFindClustersParams( seu1) table( seu1@ meta.data$ res.0.4)
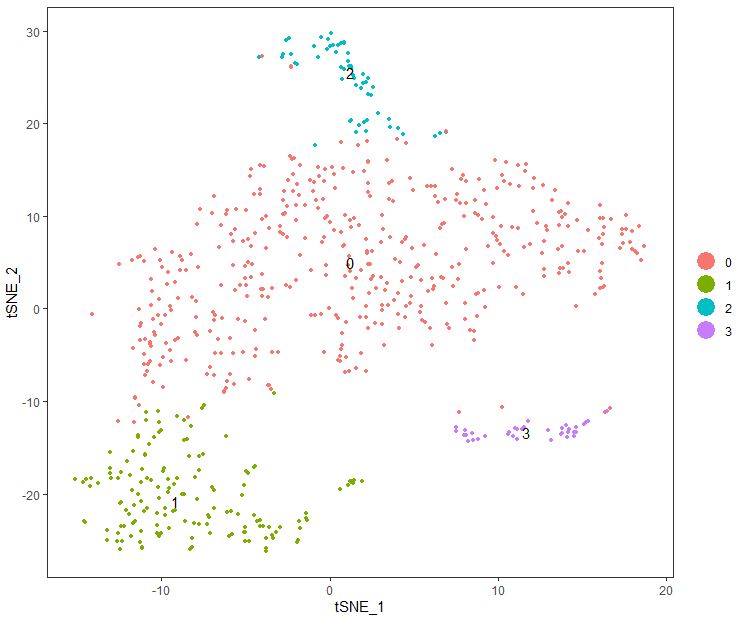
tSNE展示分组信息 1 2 3 4 5 seu= seu1 seu <- RunTSNE( object = seu, dims.use = 1 : 10 , do.fast = TRUE ) TSNEPlot( object = seu, do.label= T )
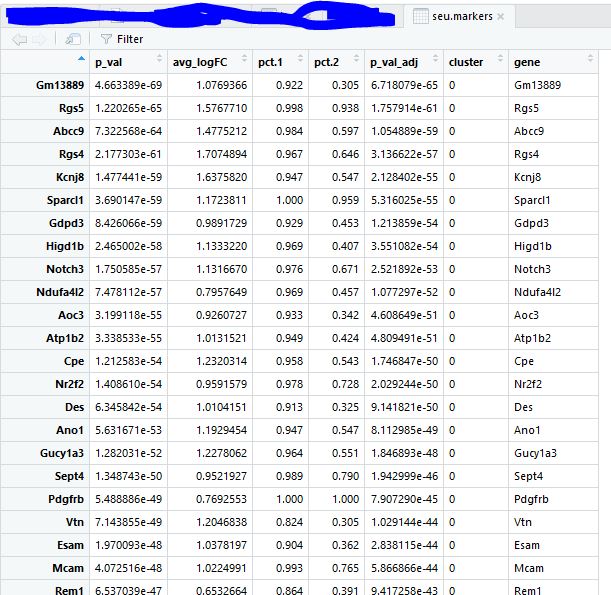
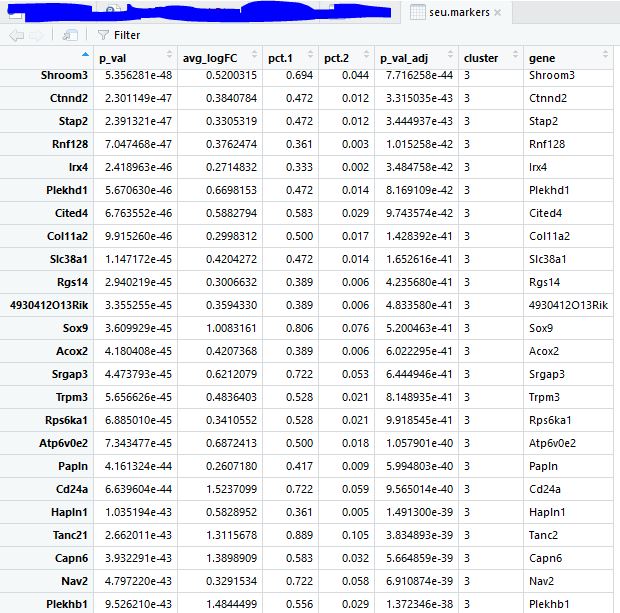
对每个类别的细胞寻找marker基因 1 2 3 seu.markers <- FindAllMarkers( object = seu, only.pos = TRUE , min.pct = 0.25 , thresh.use = 0.25 )
1 2 3 4 5 6 library( dplyr) top20 <- seu.markers %>% group_by( cluster) %>% top_n( 20 , avg_logFC) gs= read.table( 'top18-genes-in-4-subgroup.txt' ) [ , 1 ] gs intersect( top10$ gene, gs)
这时,你会发现用Seurat方法计算得到的差异基因和作者计算(自己写函数,并没有用到任何R包,有点搞不懂他为什么要这么做,还有一点小小的敬佩)得到的差异基因部分重合(37个共同基因)。 用不同的方法计算得到的差异基因都会有一定的差异,就转录组差异分析而言,limma、DESeq2、EdgeR三大差异分析包计算的差异基因也是部分重合的。 生信分析在一定程度上给研究者提供了方向,具体验证还是要落实到实验上。但是,干数据也很重要,没有方向怎么能行!所以呀,干数据和湿数据都要要,一个都不能少!
1 2 3 4 top20 <- seu.markers %>% group_by( cluster) %>% top_n( 20 , avg_logFC) intersect( top20$ gene, gs) DoHeatmap( object = seu, genes.use = top20$ gene, slim.col.label = TRUE , remove.key = TRUE )
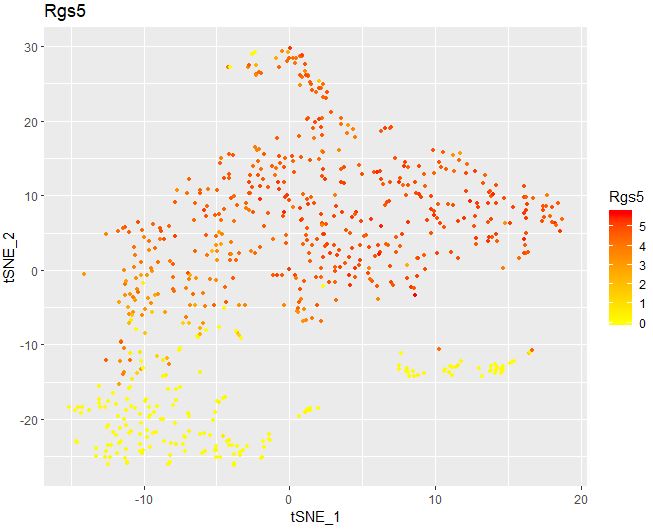
1 2 3 4 5 6 FeaturePlot( object = seu, features.plot = "Rgs5" , cols.use = c ( "yellow" , "red" ) , reduction.use = "tsne" , no.legend= FALSE )
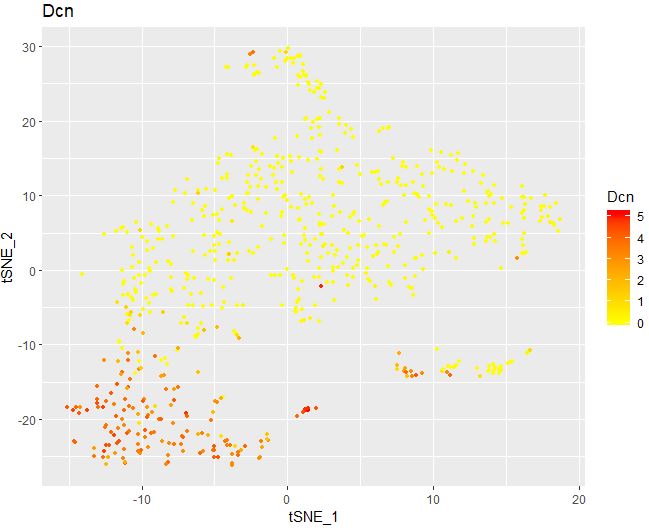
1 2 3 4 5 6 FeaturePlot( object = seu, features.plot = "Dcn" , cols.use = c ( "yellow" , "red" ) , reduction.use = "tsne" , no.legend= FALSE )
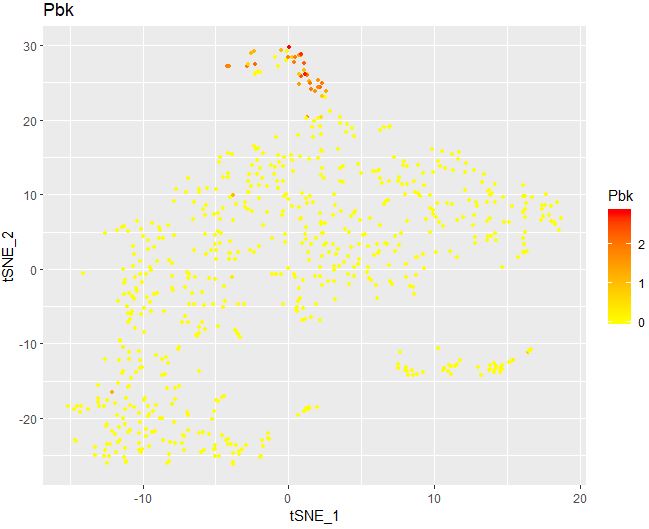
1 2 3 4 5 6 FeaturePlot( object = seu, features.plot = "Pbk" , cols.use = c ( "yellow" , "red" ) , reduction.use = "tsne" , no.legend= FALSE )
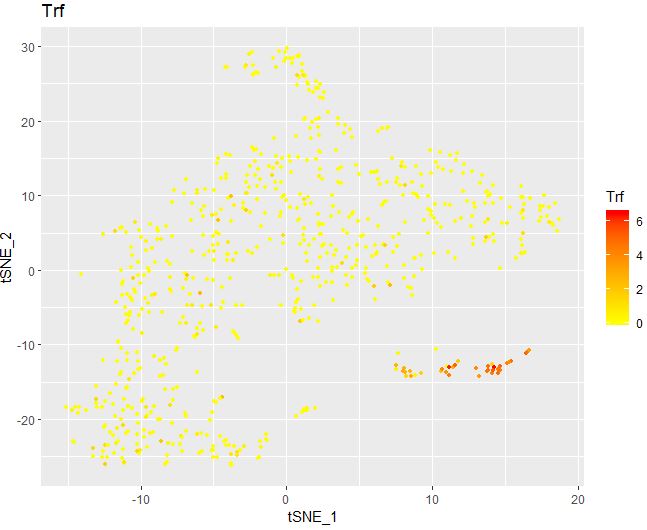
1 2 3 4 5 6 FeaturePlot( object = seu, features.plot = "Trf" , cols.use = c ( "yellow" , "red" ) , reduction.use = "tsne" , no.legend= FALSE )

 再次证明没有批次效应
再次证明没有批次效应