学习R包scater对scRNA-seq数据进行分析
scRNA-seq数据分析(二)
1
2
| rm(list = ls())
options(warn=-1)
|
认识scater
创建scater要求的对象
1
2
3
4
5
6
7
8
9
10
11
12
| library(scRNAseq)
data(fluidigm)
assay(fluidigm)<-assays(fluidigm)$rsem_counts
ct<-floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
table(rownames(ct)==0)
sample_ann<-as.data.frame(colData(fluidigm))
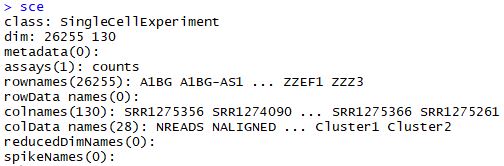
sce<-SingleCellExperiment(
assays=list(counts=ct),
colData=sample_ann
)
|
过滤
基因层面的过滤
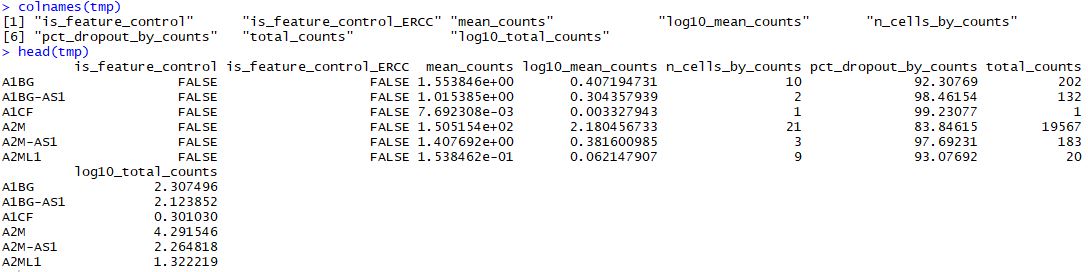
使用calculateQCMetrics函数作用于sce这个单细胞数据对象后,就可以用rowData(object)查看各个基因各项统计指标:
mean_counts:平均表达量log10_mean_counts:归一化 log10-scalepct_dropout_by_counts:该基因丢失率n_cell_by_counts:多少个细胞表达了该基因
上面的指标可以用来过滤,也可以自己计算这些统计学指标
主要是过滤掉低表达量的基因,还有线粒体基因和ERCC spike-ins的控制
1
2
3
4
5
6
7
8
9
| exprs(sce)<-log2(calculateCPM(sce)+1)
genes<-rownames(rowData(sce))
genes[grepl('^MT-',genes)]
genes[grepl('^ERCC-',genes)]
sce<-calculateQCMetrics(sce,
feature_controls = list(ERCC=grep('^ERCC',genes)))
|

1
2
3
4
|
tmp<-as.data.frame(rowData(sce))
colnames(tmp)
head(tmp)
|
1
2
3
4
5
6
|
keep_feature<-rowSums(exprs(sce)>0)>0
table(keep_feature)
sce<-sce[keep_feature,]
sce
|

细胞层面的过滤
用colData(object)可以查看各个样本统计情况
1
2
3
4
5
6
7
8
9
10
11
| tmp<-as.data.frame(colData(sce))
colnames(tmp)
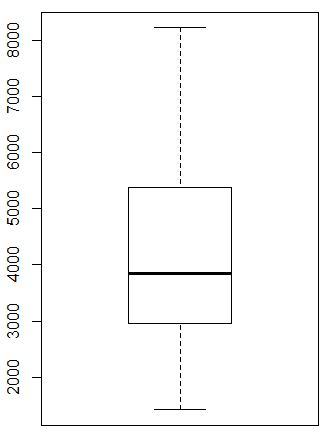
tf<-sce$total_features_by_counts
boxplot(tf)
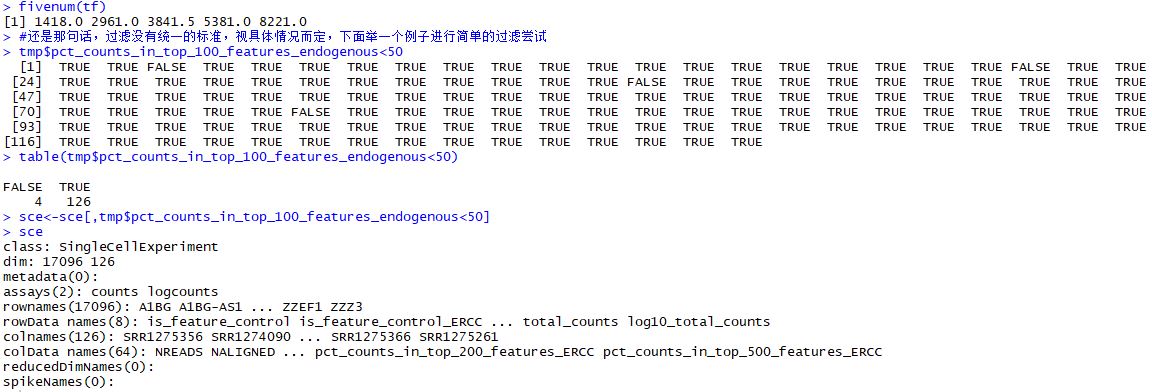
fivenum(tf)
tmp$pct_counts_in_top_100_features_endogenous<50
table(tmp$pct_counts_in_top_100_features_endogenous<50)
sce<-sce[,tmp$pct_counts_in_top_100_features_endogenous<50]
sce
|
这个sce真的是非常方便了,虽然是个list,但是却可以像dataframe一样对其进行处理,
至此,我们过滤掉了9000多个基因,4个样本
数据可视化
1
2
3
4
|
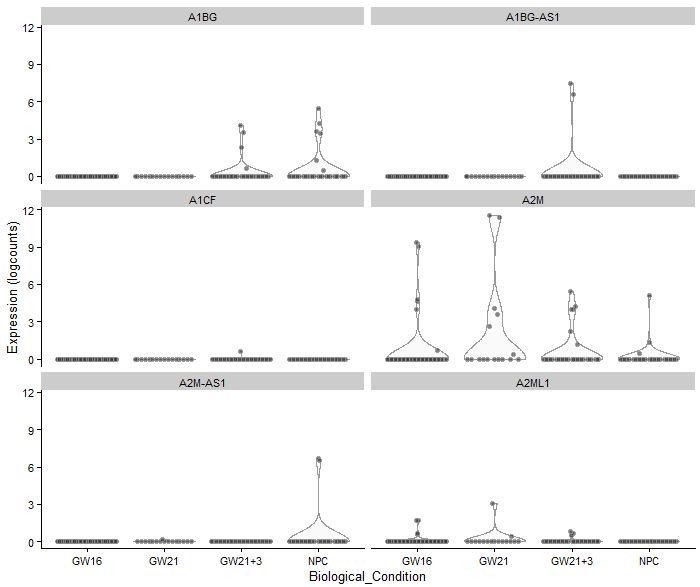
plotExpression(sce,rownames(sce)[1:6],
x="Biological_Condition",
exprs_values = "logcounts")
|
PCA
1
2
3
4
5
6
|
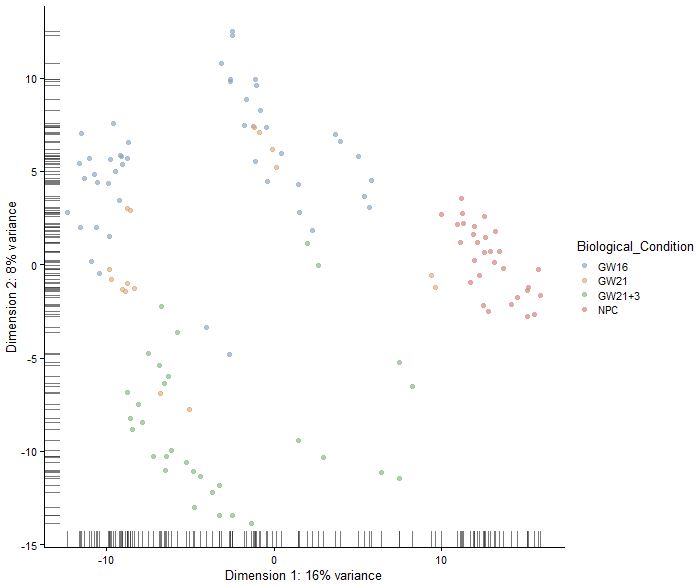
sce<-runPCA(sce)
reducedDimNames(sce)
plotReducedDim(sce,use_dimred = "PCA",colour_by = "Biological_Condition")
|
1
2
|
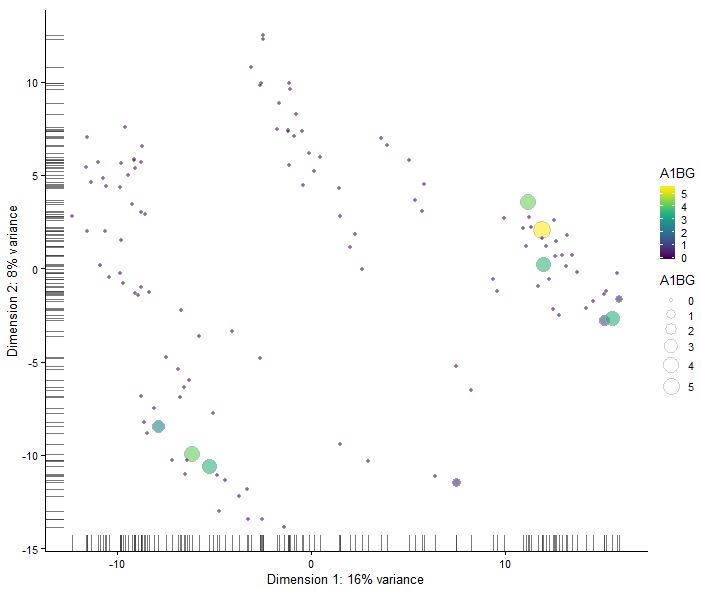
plotReducedDim(sce,use_dimred = "PCA",colour_by = rownames(sce)[1],size_by = rownames(sce)[1])
|
1
2
3
|
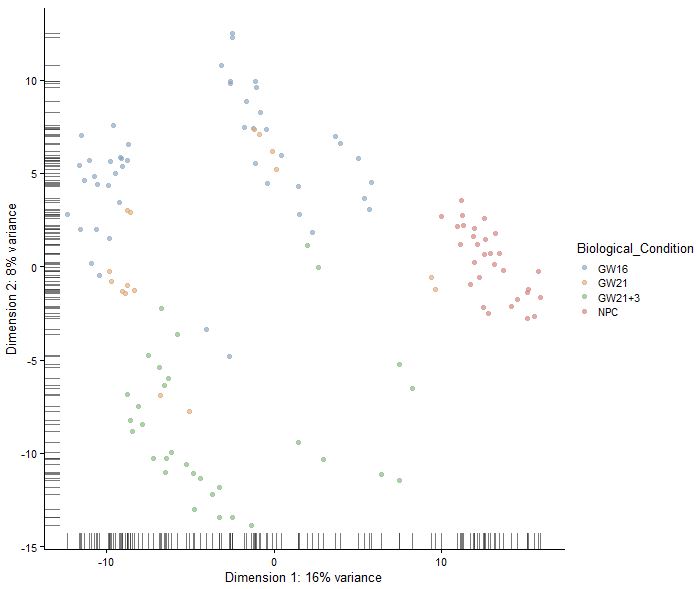
sce<-runPCA(sce,ncomponents = 20)
plotPCA(sce,colour_by="Biological_Condition")
|
1
2
|
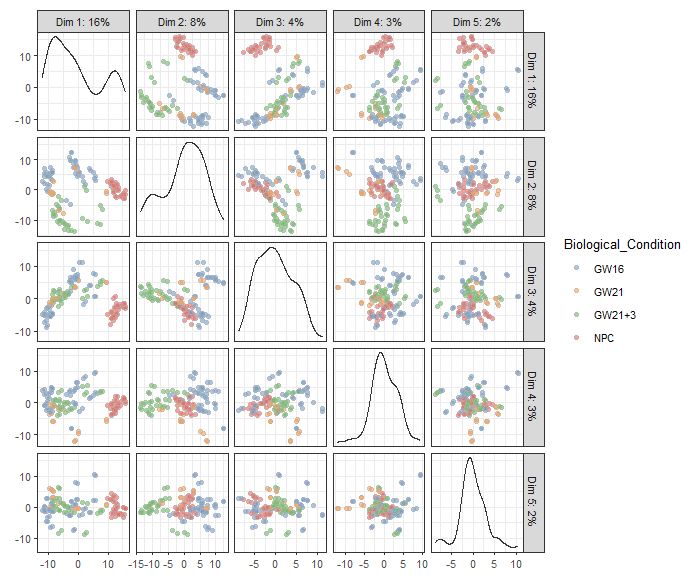
plotPCA(sce,ncomponents=5,colour_by="Biological_Condition")
|
tSNE
1
2
3
4
|
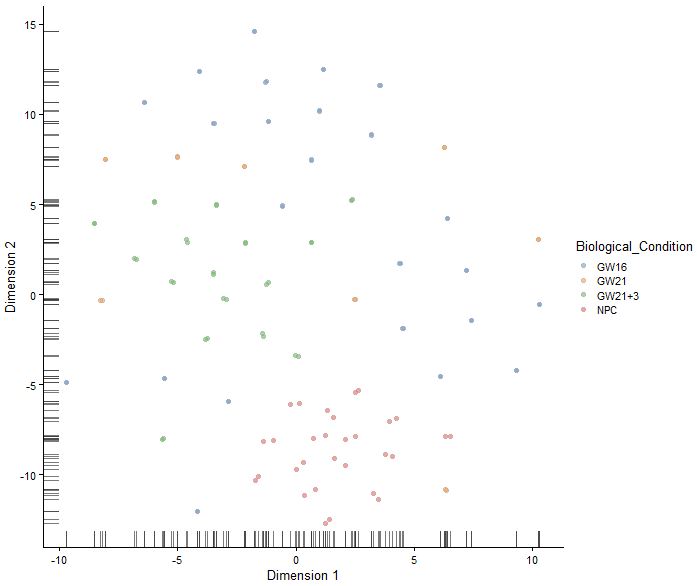
set.seed(1234)
sce<-runTSNE(sce,perplexity = 30)
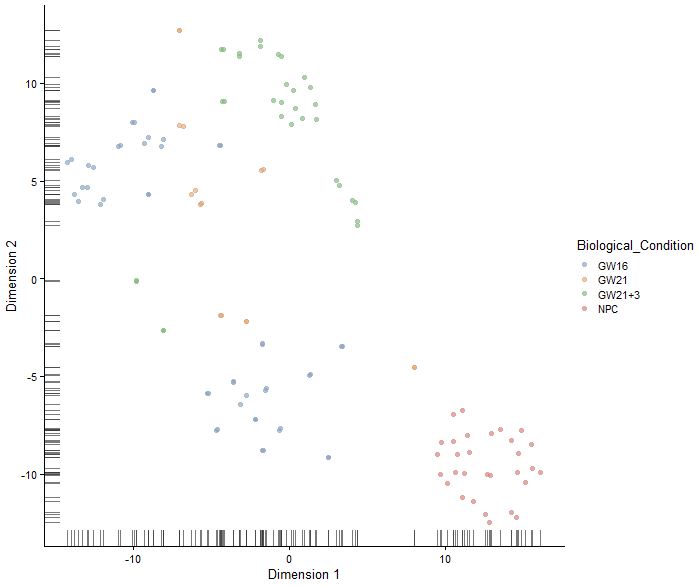
plotTSNE(sce,colour_by="Biological_Condition")
|
1
2
3
4
5
6
7
|
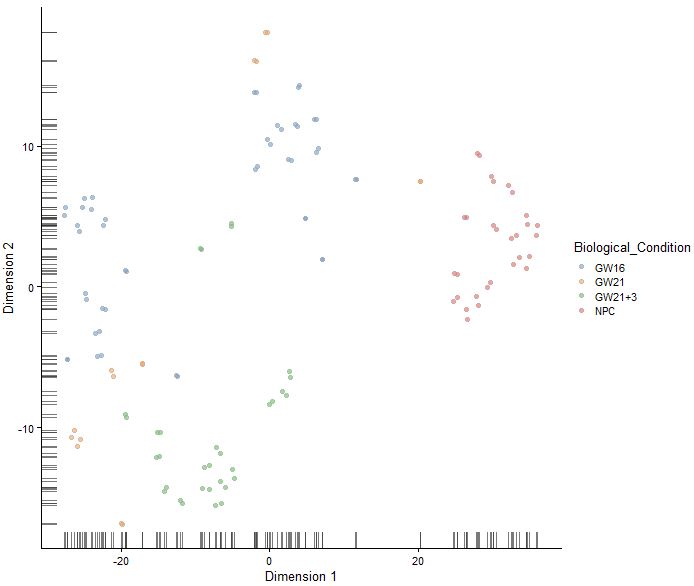
sce<-runTSNE(sce,perplexity = 10,use_dimred = "PCA",n_dimred = 10)
plotTSNE(sce,colour_by="Biological_Condition")
sce<-runTSNE(sce,perplexity = 20,use_dimred = "PCA",n_dimred = 10)
plotTSNE(sce,colour_by="Biological_Condition")
|
认识SC3
R包SC3处理scRNAseq内置的数据
1
2
3
4
5
6
7
8
9
10
11
|
library(SC3)
sce<-sce_back
sce<-sc3_estimate_k(sce)
metadata(sce)$sc3$k_estimation
rowData(sce)$feature_symbol<-rownames(rowData(sce))
kn<-4
sc3_cluster<-"sc3_4_clusters"
sce<-sc3(sce,ks=kn,biology = TRUE)
|
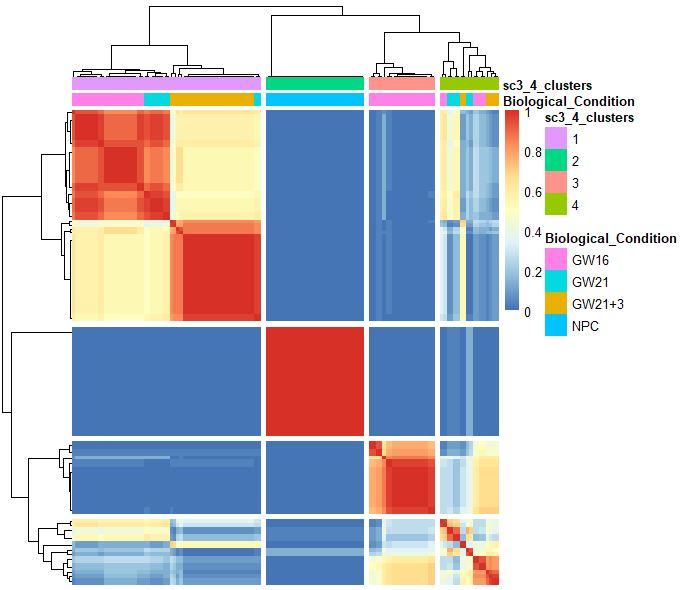
可视化展示
kn就是聚类数
1
2
|
sc3_plot_consensus(sce,k=kn,show_pdata = c("Biological_Condition",sc3_cluster))
|
1
2
|
sc3_plot_expression(sce,k=kn,show_pdata = c("Biological_Condition",sc3_cluster))
|
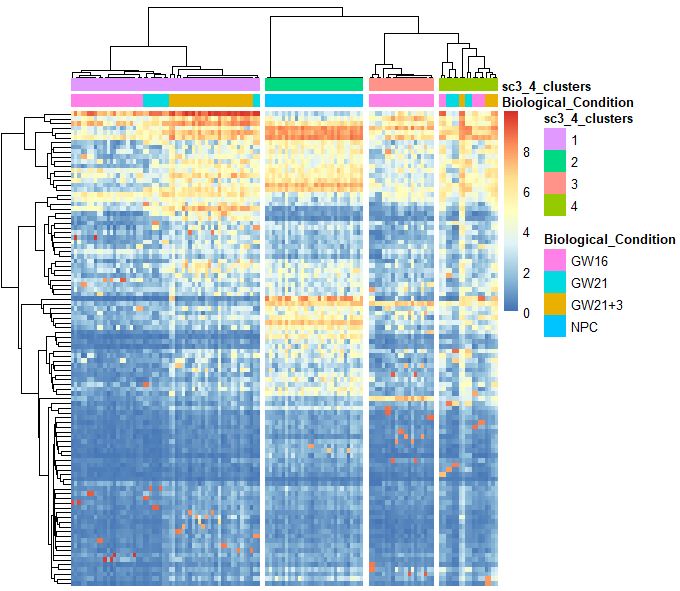
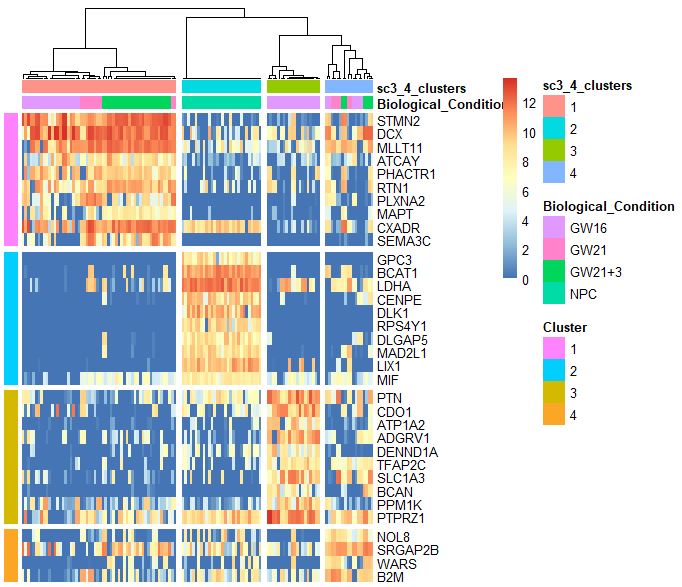
SC3包找marker基因
1
2
|
sc3_plot_markers(sce,k=kn,show_pdata =c("Biological_Condition",sc3_cluster) )
|