···
scRNA-seq数据分析流程
以下分析流程以GSE111229数据为例(为什么用它呢,因为它数据量小呀,嘿嘿🤭)在进行分析之前,首先了解一下该套数据的基本情况。
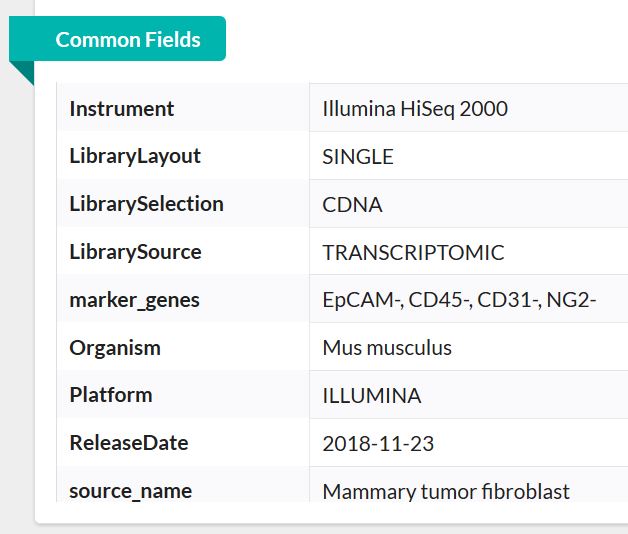
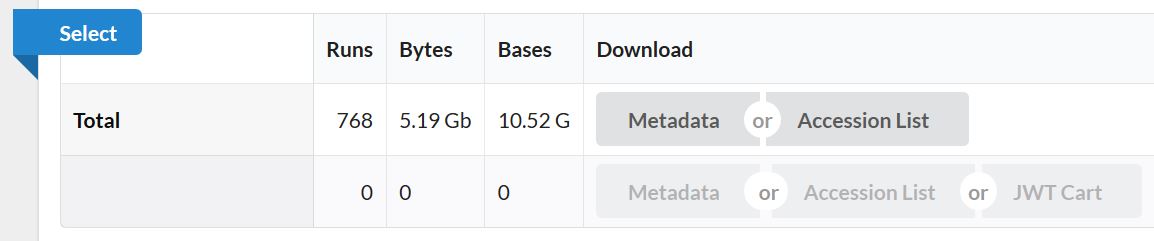
由此可基本了解,此数据为单端测序数据,物种为小鼠。768个样本即768个单细胞测序数据,总大小才10G。这在单细胞测序数据中真的是非常小!非常罕见啦!
😄数据分析上游(Linux)😄
因为是单细胞转录组测序数据,和转录组测序的上游分析没什么区别。所以以下分析流程都是在conda构建的rna环境下进行,这个环境也是很久之前搭建好的,在此不再赘述。
运行上述代码后,可以看到提示符(prompt)左边多了一个(rna)环境,此环境里面,预先装好了处理RNA-seq数据所需要的相关软件。

数据下载
如果样本量不多,网速又不理想,推荐EBI-ENA数据库,此数据库下载网速还可以,另外该数据库除了提供SRA文件外,还提供FASTQ文件,省去了从NCBI下载SRA文件转为FASTQ文件的麻烦(如何单个文件数据量大,此格式转换步骤非常耗时)。
如果样本量特别多,网速又比较理想,推荐使用Aspera、prfetch、wget、curl。前两者大大优于后两者。Aspera Connect的下载速度是最快了,此方法也可以下载FASTQ和SRA文件,免去了SRA转至FASTQ的过程,该过程很耗时,十分耗时。
进入EBI官网,输入SRP号
1
2

3
以其中一个样本SRR6791132为例,点击SRR6791132进入下载界面,鼠标右键红色箭头处——“复制链接地址”
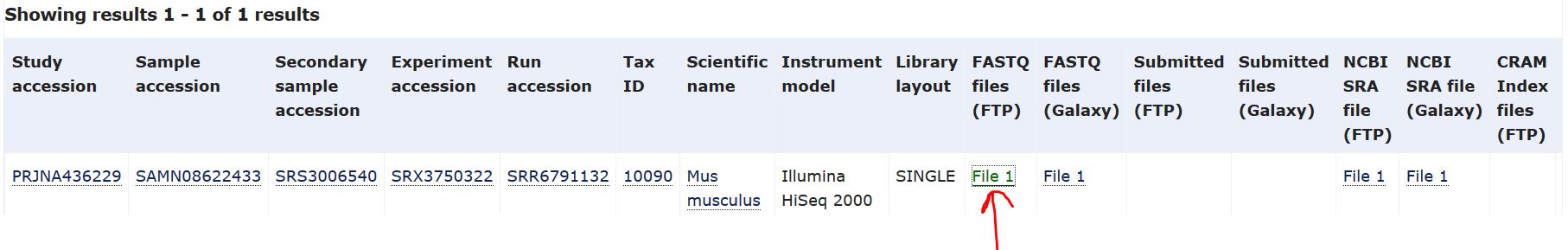
进入linux进行数据下载
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
| mkdir -p scRNA-seq/fastq && cd scRNA-seq/fastq
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/002/SRR6791132/SRR6791132.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/005/SRR6791135/SRR6791135.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR679/005/SRR6791135/SRR6791135.fastq.gz
```
<font color=blue>注意注意,特别注意,要下载整套数据最好使用Aspera或者prefetch,本宝宝没有办法呀,网不好,空间也不大,linux中的数据操作只能用三个样本做演示了,熟悉一下分析流程。如果是整套数据,记得写小循环,放后台,就不用一直守着它了,最好是睡觉的时候跑,不耽误事。</font>
```bash
mkdir -p /scRNA-seq/html && cd /scRNA-seq/html
ls /scRNA-seq/fastq/*gz|xargs fastqc -t 4 -o ./
```
上述操作会得到html文件和zip文件
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/quality.JPG"/>
网页打开其中一个html文件可查看测序数据的质量,该数据整体质量还不错([质控报告解读教程](https://www.jianshu.com/p/835fd925d6ee))
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/html.JPG" width="50%" height="50%">
大部分测序数据都会出现如下情况,不过这个问题不大
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/pbsc.JPG" width="50%" height="50%">
```bash
|
比对
1、小鼠参考基因组
首先下载小鼠参考基因组,hisat2或者bowtie2构建小鼠参考基因组索引。也可以直接下载参考基因组索引文件。另外还需要下载小鼠参考基因组注释文件。(此步骤很早前处理RNA-seq数据时就已经完成了,就不再赘述)
hisat2构建的mm10参考基因组索引文件
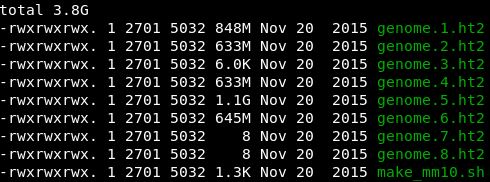
mm10注释文件

2、进行参考基因组比对
1
2
3
4
5
6
|
ls *.gz | while read id; do hisat2 -p 4 -x /addfirst/reference/mouse/index/hisat2-build-index/mm10/genome -U $id -S ${id%%.*}.hisat2.sam;done
mkdir /scRNA-seq/align && cd /scRNA-seq/align
cp /scRNA-seq/fastq/*.sam /scRNA-seq/fastq/*.bam ./
ls *.sam | while read id; do samtools sort -O bam -@ 4 -o ${id%%.*}.hisat2.bam $id; done
|
比对结果如下:82.25%的比对率,比较的凑合了,一般情况最好90%以上,后两个数据的比对率实在不能看。真实处理环境一定要仔细调节过滤参数!!!
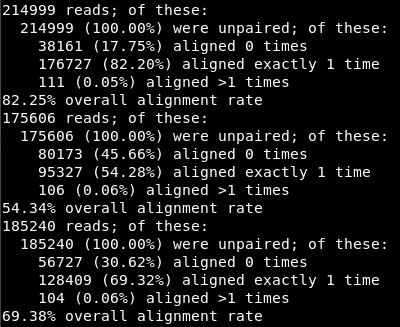
可以比较一下bam、fasatq、sam文件的大小。
sam文件远远大于bam和fastq文件。生信分析上游一定要对文件大小保持高度敏感,避免不必要的错误。另外,数据量大的话,可以在生成sam文件时通过|将结果进一步生成bam文件,这样可以大大节省存储空间。
构建index文件
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
| ls *.bam | xargs -i samtools index {}
```
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/bamindex.JPG"/>
```bash
gtf=/addfirst/reference/mouse/gencode.vM23.annotation.gtf
featureCounts -T 4 -p -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1
查看counts矩阵
less -S all.id.txt
```
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/reads.JPG"/>
查看counts矩阵会发现绝大多数基因表达量都是0,因为对于大多数细胞来说,大多数基因都是测不到的,这个属于正常现象。在此,我们只用了3个样本做演示,总数据则有768个样本。
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/countsmatrix.JPG"/>
理论上按照上面的分析流程生成的表达矩阵数据和下图中NCBI官网上的rawCounts.txt.gz的内容应该是差不多的,只是所用处理软件不同会有一点点区别。
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/NCBImatrix.JPG"/>
至此,scRNA-seq上游分析结束,这里比较重要的是**数据质量**,数据质量直接决定比对的效率,影响最终生成的表达矩阵的可靠性。所以质控步骤要严格把控,送样测序也要选靠谱的公司。
如果是使用rawCounts数据进行下游分析,首先要对数据进行标准化,然后再做后续分析。该篇文章是使用rpkm的数据进行下游分析,这里也直接使用rpkm的数据进行下游分析。
```R
a=read.table('../GSE111229_Mammary_Tumor_fibroblasts_768samples_rpkmNormalized.txt.gz',header = T ,sep = '\t')
a[1:6,1:4]
dat=a[apply(a,1, function(x) sum(x>0) > floor(ncol(a)/50)),]
dat[1:4,1:4]
```
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/a.JPG"/>
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/dat.JPG"/>
```R
hc=hclust(dist(t(log(dat+0.1))))
plot(hc,labels = F)
clus = cutree(hc, 4)
group_list= as.factor(clus)
table(group_list)
```
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/hclust.JPG" width="60%" height="60%">
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/grouplist.JPG"/>
```R
colnames(dat)
library(stringr)
plate=str_split(colnames(dat),'_',simplify = T)[,3]
table(plate)
n_g = apply(a,2,function(x) sum(x>0))
df=data.frame(g=group_list,plate=plate,n_g=n_g)
df$all='all'
metadata=df
df[1:4,1:4]
save(dat,metadata,file = '../input_rpkm.Rdata')
|
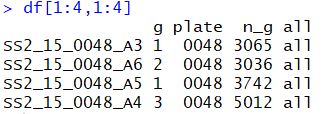
检测是否存在批次效应
因为该篇文章使用两块384孔进行测序,所以要先确定两块板是否存在批次效应,如果存在,则两块板的样本不能合并分析,如果不存在批次效应,则两块板可以进行批次分析。
PCA主成份分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
| rm(list = ls())
options(stringsAsFactors = F)
load(file = '../input_rpkm.Rdata')
dat[1:4,1:4]
head(metadata)
plate=metadata$plate
dat_back=dat
dat=t(dat)
dat=as.data.frame(dat)
dat=cbind(dat,plate )
dat[1:4,12688:12690]
table(dat$plate)
library("FactoMineR")
library("factoextra")
dat.pca <- PCA(dat[,-ncol(dat)], graph = FALSE)
fviz_pca_ind(dat.pca,
col.ind = dat$plate,
addEllipses = FALSE,
legend.title = "Groups"
)
```
<img src="https://blog-image-host.oss-cn-shanghai.aliyuncs.com/gyqblog/PCA.jpeg" width="60%" height="60%">
**tSNE分析**
```R
rm(list = ls())
options(stringsAsFactors = F)
library(Rtsne)
load(file = '../input_rpkm.Rdata')
dat[1:4,1:4]
dat_matrix <- t(dat)
dat_matrix=log2(dat_matrix+0.01)
set.seed(42)
tsne_out <- Rtsne(dat_matrix,pca=FALSE,perplexity=30,theta=0.0)
tsnes=tsne_out$Y
tsnes=as.data.frame(tsnes)
group=c(rep('plate1',384),rep('plate2',384))
colnames(tsnes)<-c("tSNE1","tSNE2")
library(ggfortify)
ggplot(tsnes, aes(x = tSNE1, y = tSNE2))+ geom_point(aes(col=group))+ theme_classic()
|
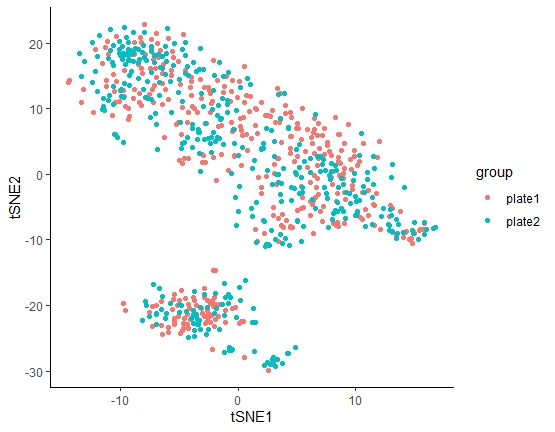
细胞亚群
#########################
##针对所有细胞做PCA分析##
#########################
rm(list = ls()) ## 魔幻操作,一键清空
options(stringsAsFactors = F)
load(file = '../input_rpkm.Rdata')
dat[1:4,1:4]
head(metadata)
group<-metadata$g
#做好数据备份
dat_back=dat
dat=t(dat) #记得转置数据
dat=as.data.frame(dat)
dat=cbind(dat,group ) #cbind根据列进行合并,即叠加所有列,为矩阵添加分组信息
dat[1:4,12688:12690]
table(dat$group)
library("FactoMineR")
library("factoextra")
dat.pca <- PCA(dat[,-ncol(dat)], graph = FALSE)
fviz_pca_ind(dat.pca,#repel =T,
geom.ind = "point", # show points only (nbut not "text")
col.ind = dat$group, # color by groups
addEllipses = FALSE, # Concentration ellipses
legend.title = "Groups"
)
#########################
##针对所有细胞做tSNE分析##
#########################
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(Rtsne)
load(file = '../input_rpkm.Rdata')
dat[1:4,1:4]
dat_matrix <- t(dat)
dat_matrix=log2(dat_matrix+0.01)
# Set a seed if you want reproducible results
set.seed(42)
tsne_out <- Rtsne(dat_matrix,pca=FALSE,perplexity=30,theta=0.0)
group=metadata$g
##添加颜色
tsnes=tsne_out$Y
tsnes=as.data.frame(tsnes)
colnames(tsnes)<-c("tSNE1","tSNE2")
ggplot(tsnes, aes(x = tSNE1, y = tSNE2))+ geom_point(aes(col=group))+ theme_classic()
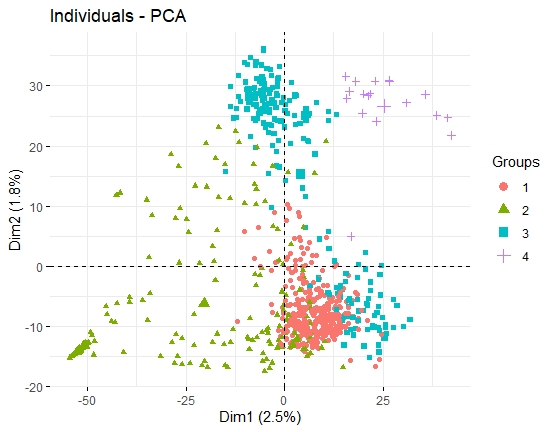

单细胞测序分析旨在分出不同细胞亚群(展现细胞间的异质性),力求寻找某类亚群与某特定生物学功能之间的联系。而要探讨细胞分群,就必须要知道PCA和tSNE。
PCA(Principal Component Analysis)
tSNE(t-Distributed Stochastic Neighbor Embedding)
什么样的算法才是最理想的能够将细胞分群的算法呢?
1)局部结构:属于同一个亚群的细胞,聚类尽可能近
2)全局结构:属于不同亚群的细胞,聚类尽可能分来
- PCA的方法侧重于去抓样本中隐含的主要效应,从而让差异大的样本在图上呈现较远的距离。常规RNA-seq项目中,处理效应、批次效应、离群效应等属于较大的效应,PCA的方法可以良好的去展示这些效应。
- 如果影响样本分组的不是主要效应,而是一些更小的效应,PCA则无法对样本进行准确的区分。scRNA-seq的可视化主要期望对各个细胞亚群有良好的区分。每次检测的上万甚至几十万个细胞中,几乎肯定可以区分出几十中细胞亚群,包括一些稀有的细胞。而区分这些细胞亚群(尤其是稀有细胞类型)的效应,往往不是主要效应(即大量基因的差异),而是一些微小效应(少量标记基因差异)。如果使用PCA进行分析的话,就会掩盖掉某些微小的但是有可能非常重要的细胞群。
- tSNE算法就属于可以同时兼顾局部结构和全局结构的非线性降维可视化算法。tSNE不同于PCA(PCA主要目标是尽量去抓取群体中的主要效益),tSNE算法的主要目标是:降维后的数据依然保持最为相似的紧密成簇。这就保证了哪怕某些稀有细胞只是少量基因区别于其他细胞亚群,在tSNE中依然可以与其他细胞有良好的区分。
结论:PCA是常规转录组常用的数据降维和样本关系可视化的方法,但针对群体单细胞转录组数据,tSNE是明显胜过PCA的方法!
参考资料
http://www.sohu.com/a/320164491_278730