whole exome-sequencing analysis pipeline
全外显子数据分析 (一) 环境搭建 1、GATK依赖Java 8/JDK 1.8 (Oracle or OpenJDK)
2、下载安装GATK4
1 2 3 4 5 wget https://github.com/broadinstitute/gatk/releases/download/4.2.0.0/gatk-4.2.0.0.zip unzip gatk-4.2.0.0.zip echo 'export PATH=/home/gongyuqi/biosoft/GATK/gatk-4.2.0.0:$PATH' >> ~/.bashrcsource ~/.bashrc
3、下载其他需要的软件
1 2 3 4 5 6 7 8 9 10 11 12 conda create -n WES conda install -y python=3.6.2 conda install -y bwa sra-tools samtools bcftools snpEFF multiqc qualimap conda activate WES cd /home/gongyuqi/project/WESmkdir raw qc clean mutaioncd qc && mkdir raw_qc clean_qc
(二) WES测试数据下载 1、数据来源GSE153707 ,我们这里从EBI直接下载fastq文件
1 2 3 4 5 6 7 8 9 10 11 12 13 dir =/home/gongyuqi/.aspera/connect/etc/asperaweb_id_dsa.opensshx=_1 y=_2 for id in {11,12}do ascp -QT -l 300m -P33001 -i $dir era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR121/0$id /SRR121359$id /SRR121359$id $x .fastq.gz . ascp -QT -l 300m -P33001 -i $dir era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR121/0$id /SRR121359$id /SRR121359$id $y .fastq.gz . done chmod +x download.shnohup ./download.sh &
2、了解文章WES数据处理的相关步骤和参数
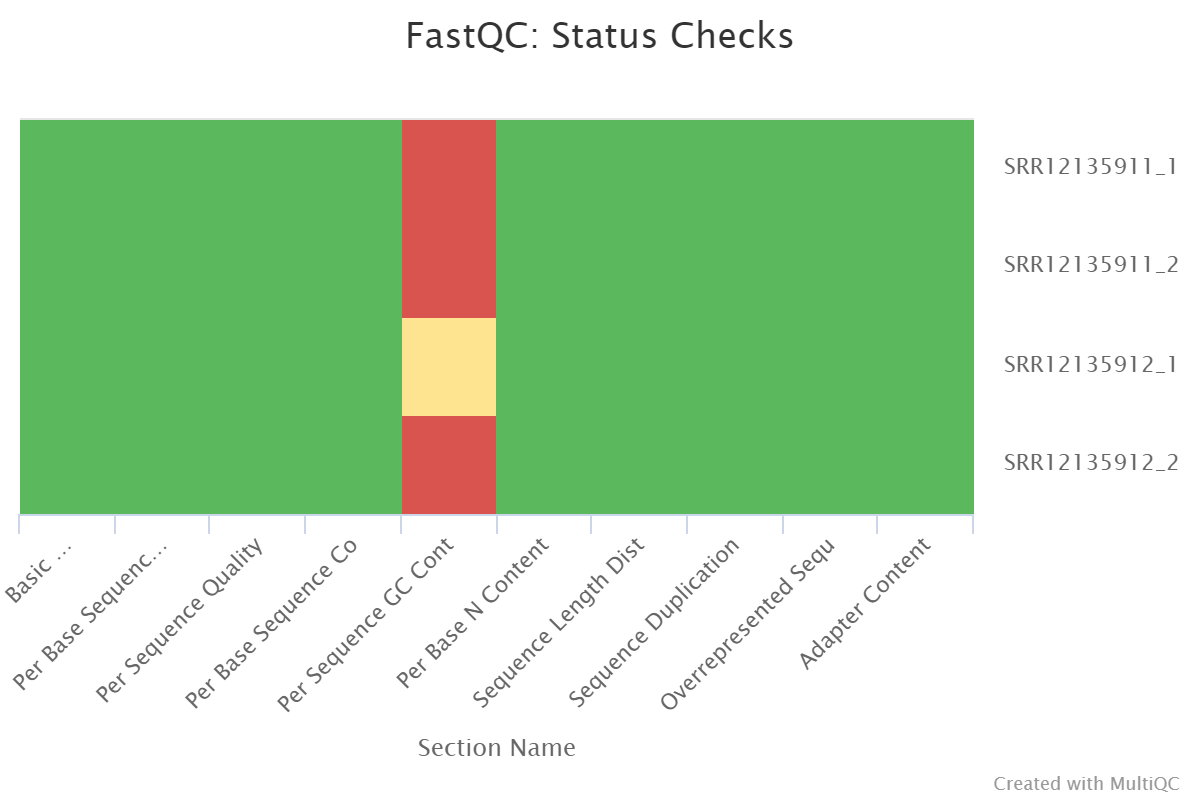
(三) GATK准备bam文件用于找变异 1、 比对GATK官网提供的参考基因组 1 2 3 4 ls ../raw/*.gz|xargs fastqc -t 4 -o ./multiqc *.zip
1 2 3 4 5 6 7 8 9 10 11 12 13 ls *1.fastq.gz >fq1.txtls *2.fastq.gz >fq2.txtpaste fq1.txt fq2.txt > sample.txtindex=/home/gongyuqi/ref/GATK/hg38/v0/Homo_sapiens_assembly38.fasta.64 cat sample.txt | while read id do nohup arr=(${id} ) fq1=${arr[0]} fq2=${arr[1]} sample=${fq1%%_*} bwa mem -t 8 -R "@RG\tID:$sample \tSM:$sample \tLB:WGS\tPL:Illumina" /$index $fq1 $fq2 |samtools sort -@ 8 -o ../align/$sample .sort.bam - &>>../align/$sample .log & done
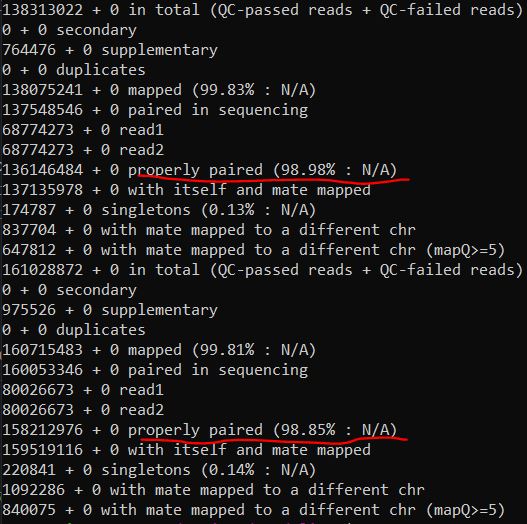
1 2 ls *.bam|while read id ;do samtools flagstat $id ;done
2、标记PCR重复 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 ls *.bam|while read sampledo nohup gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" \ MarkDuplicates \ --INPUT $sample \ --OUTPUT ${sample%%.*} _marked.bam \ -M ${sample%%.*} .metrics & done ls *_marked.bam|while read sampledo nohup gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" \ FixMateInformation \ -I ${sample} \ -O ${sample%%_*} _marked_fixed.bam \ -SO coordinate & done ls *_fixed.bam|while read sampledo nohup samtools index $sample &done
3、碱基矫正 BaseRecalibrator 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 ref=/home/gongyuqi/ref/GATK/hg38/v0/Homo_sapiens_assembly38.fasta snp=/home/gongyuqi/ref/GATK/hg38/v0/Homo_sapiens_assembly38.dbsnp138.vcf indel=/home/gongyuqi/ref/GATK/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz ls *_marked_fixed.bam|while read sampledo nohup gatk \ --java-options "-Xmx20G -Djava.io.tmpdir=./" \ BaseRecalibrator \ -R $ref \ -I $sample \ --known-sites $snp \ --known-sites $indel \ -O ${sample%%_*} _recal.table \ 1>${sample%%_*} _log.recal 2>&1 & done ls *_marked_fixed.bam|while read sampledo nohup gatk \ --java-options "-Xmx20G -Djava.io.tmpdir=./" ApplyBQSR \ -R $ref \ -I $sample \ -bqsr ${sample%%_*} _recal.table \ -O ${sample%%_*} _bqsr.bam \ 1>${sample%%_*} _log.ApplyBQSR 2>&1 & done ls *bqsr.bam|while read id do nohup samtools flagstat $id &done gatk --java-options "-Xmx20G" AnalyzeCovariates \ -before recal_data.table1 \ -after recal_data.table2 \ -plots AnalyzeCovariates.pdf
4、HaplotypeCaller
这一步是使用GATK的HaplotypeCaller找变异,但现在GATK官网推荐GATK4+Mutect2找变异,在这里我们还是简单运行一下HaplotypeCaller。
1 2 3 4 5 6 7 8 9 ls *bqsr.bam|while read sampledo nohup gatk \ --java-options "-Xmx20G -Djava.io.tmpdir=./" HaplotypeCaller \ -R $ref \ -I $sample \ --dbsnp $snp \ -O ../vcf/${sample%%_*} _raw.vcf \ 1>../vcf/${sample%%_*} _log.HC 2>&1 & done
(三) Mutect2找变异 Mutect2 v4.1.0.0的教程 目前已经不被GATK官方强烈推荐。官方更新推荐了Mutect2 v4.1.1.0 and later版本的使用教程 ,更新过的教程比较推荐用于正常对照样本>40的WES测序数据。如果没有这么多的样本量,Mutect2 v4.1.0.0的教程也同样可以使用,只是有部分参数在新的Mutect2版本中被弃用或是将其功能整合到另外的参数去了。具体问题可以查看官网说明。
1、过滤种系突变
首先需要一个germline variant sites VCF文件,去官网下载af-only-gnomad.hg38.vcf.gz 文件。记得一并下载对应的.tbi文件
1 2 3 4 5 6 7 8 9 10 11 12 13 14 ref=/home/gongyuqi/ref/GATK/hg38/v0/Homo_sapiens_assembly38.fasta germine_vcf=/home/gongyuqi/ref/GATK/hg38/af-only-gnomad/af-only-gnomad.hg38.vcf.gz nohup gatk --java-options "-Xmx20g" Mutect2 \-R $ref \ -I SRR12135912_bqsr.bam \ -tumor SRR12135912 \ -I SRR12135911_bqsr.bam \ -normal SRR12135911 \ --germline-resource $germine_vcf \ --af-of-alleles-not-in-resource 0.0000025 \ --disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \ --bam-output HQ461-untreated-Mutect2.bam \ -O ../Mutect2/HQ461-untreated.Mutect2.vcf &
生成文件如下:
2、考虑样品纯度及假阳性
通常,病人的肿瘤样本是混合有正常组织的,这个时候我们可以选择GetPileupSummaries工具来计算肿瘤样本的污染程度。对于Mutect2流程而言,GATK只考虑了肿瘤样本中正常样本的污染情况,不考虑正常样本中肿瘤样本的污染情况。这里我们的处理组:HQ461,对照组untreated。,虽然我们的样本是肿瘤细胞的单克隆株,我们这里还是跑一下这个流程。
step 1、生成af-only-gnomad.hg38.SNP_biallelic.vcf.gz 1 2 3 4 5 6 7 8 ref=/home/gongyuqi/ref/GATK/hg38/v0/Homo_sapiens_assembly38.fasta germine_vcf=/home/gongyuqi/ref/GATK/hg38/af-only-gnomad/af-only-gnomad.hg38.vcf.gz nohup gatk SelectVariants \-R $ref \ -V $germine_vcf \ --select-type-to-include SNP \ --restrict-alleles-to BIALLELIC \ -O /home/gongyuqi/ref/GATK/hg38/af-only-gnomad/af-only-gnomad.hg38.SNP_biallelic.vcf.gz &
step 2、GetPileupSummaries 1 2 3 4 5 6 7 8 9 10 11 12 13 14 germine_biallelic_vcf=/home/gongyuqi/ref/GATK/hg38/af-only-gnomad/af-only-gnomad.hg38.SNP_biallelic.vcf.gz genomic_intervals=/home/gongyuqi/ref/GATK/hg38/v0/wgs_calling_regions.hg38.interval_list nohup gatk GetPileupSummaries \-I SRR12135911_bqsr.bam \ -L $genomic_intervals \ -V $germine_biallelic_vcf \ -O ../Mutect2/SRR12135911.pileups.table & nohup gatk GetPileupSummaries \-I SRR12135912_bqsr.bam \ -L $genomic_intervals \ -V $germine_biallelic_vcf \ -O ../Mutect2/SRR12135912.pileups.table &
step 3、 CalculateContamination
计算污染比例,用以过滤掉somatic variant中一些可能由污染导致的假阳性突变
1 2 3 4 nohup gatk CalculateContamination \-I SRR12135912.pileups.table \ -matched SRR12135911.pileups.table \ -O HQ461-untreated.calculatecontamination.table &
step 3、 FilterMutectCalls
结合CalculateContamination的评估,进行最后的过滤
1 2 3 4 5 6 ref=/home/gongyuqi/ref/GATK/hg38/v0/Homo_sapiens_assembly38.fasta nohup gatk FilterMutectCalls \-R $ref \ -V HQ461-untreated.Mutect2.vcf \ --contamination-table HQ461-untreated.calculatecontamination.table \ -O HQ461-untreated.filtered.Mutect2.vcf &
至此,我们找变异的步骤就差不多走完了。你会发现我们并没有使用到CollectSequencingArtifactMetrics和FilterByOrientationBias参数,那是因为我们当前版本的Mutect2已经将FilterByOrientationBias的功能整合进FilterMutectCalls中了,详情见GATK论坛中的一篇帖子 。
另外,在做WES分析时,对照组是很重要的。拿肿瘤样本举例,tumor-only的模式会得到很多很多很多的假阳性,详情见GATK论坛中的另一篇帖子 。对照组的存在会大大降低假阳性率。
(四) SnpEff & SnpSift注释
以下流程参考SnpEff & SnpSift官网文档
1、SnpEff软件及所需注释数据库的下载 step 1、下载安装SnpEff软件 下载SnpEff ,现在SnpEff和SnpSift是绑定在一起的。
1 2 3 4 5 cd ~/biosoft/wget https://snpeff.blob.core.windows.net/versions/snpEff_latest_core.zip & unzip snpEff_latest_core.zip
step 2、下载SnpEff软件需要的数据库文件 下载 SnpEff databases: 官网给的命令是java -jar snpEff.jar download GRCh38.76。但是运行这个命令会报错:找不到GRCh38.76
1 2 3 4 5 java -jar snpEff.jar databases | less java -jar snpEff.jar databases | grep -i GRCh38
1 2 3 4 nohup java -jar snpEff.jar download GRCh38.99 &
GRCh38.99内容如下
2、SnpEff、SnpSift使用 step 1、SnpEff注释 1 2 java -Xmx16g -jar snpEff.jar -v GRCh38.99 /home/gongyuqi/project/WES/Mutect2/HQ461-untreated.filtered.Mutect2.vcf > HQ461-untreated.filtered.ann.vcf
step 2、SnpSift过滤 该部分同时参考官网说明文档及某优秀博客用SnpSift过滤VCF文件
1 2 3 4 cat HQ461-untreated.filtered.ann.vcf | java -jar SnpSift.jar filter "( na FILTER ) | (FILTER = 'PASS')" > HQ461-untreated.filtered.ann.snpsift.vcfcat HQ461-untreated.filtered.ann.snpsift.vcf | java -jar SnpSift.jar filter "(ANN[0].IMPACT has 'HIGH') | (ANN[0].IMPACT has 'MODERATE')" > HQ461-untreated.filtered.ann.snpsift.second.vcf
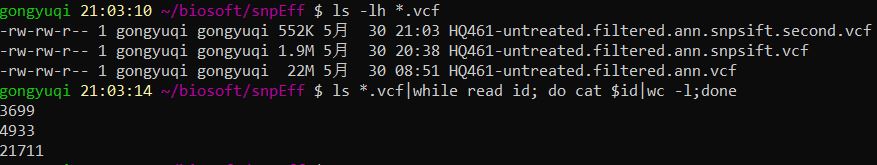
1 2 3 ls -lh *.vcfls *.vcf|while read id ; do cat $id |wc -l;done
step 3、sed命令复现SnpSift的功能
这里不难发现,SnpSift其实就是对文本文件的处理。要是shell脚本够扎实,也完全可以不依赖SnpSift。重要的是,在学习NGS组学分析过程中,但凡有锻炼自己shell脚本的地方,就一定要抓住机会动手写一写💪 这里我们可以用一个简单的sed命令解决
1 2 3 4 5 6 cat HQ461-untreated.filtered.ann.vcf | grep -v "#" | \sed -n '/PASS/p' - | \ sed -n '/\(HIGH\|MODERATE\)/p' - \ >test_PASS_HIGH_MODERATE.third.vcf
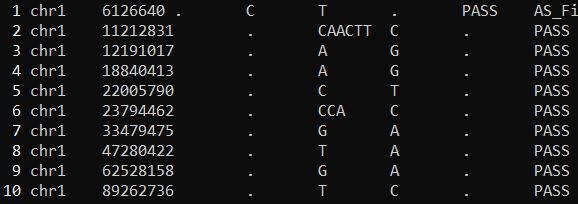
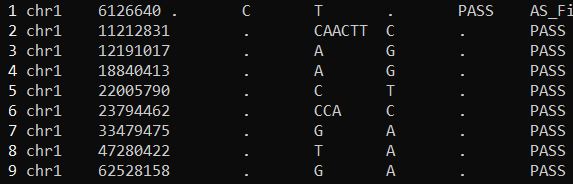
1 2 3 4 cat HQ461-untreated.filtered.ann.snpsift.second.vcf|grep -v "#" |wc -lcat test_PASS_HIGH_MODERATE.second.vcf|wc -l
1 2 3 cat HQ461-untreated.filtered.ann.snpsift.second.vcf|grep -v "#" |less -SNcat test_PASS_HIGH_MODERATE.second.vcf|less -SN
HQ461-untreated.filtered.ann.snpsift.second.vcf
(五) 原文数据复现
真正接受检验的时候到了,看看我们的pipeline能不能复现文章的数据。这也是进一步检验我们pipeline的正确性的重要环节(当然也不能迷信文章的结果,但是7+文章的数据分析质量应该还是可以作为一个较好的参考)。
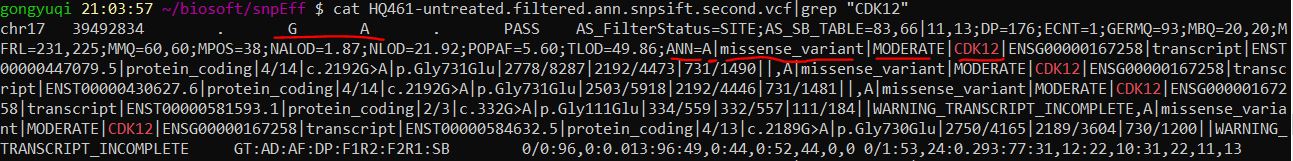
再看看我们过滤后的vcf文件中是否有检测到CDK12的G到A的突变
学习WES心得体会
1、学习某个软件的用法时,主要参考官方文档!!!可以选择性参考相关博客主博客,但切记拿来主义!!!深刻理解用到的每个参数。
2、WES的相关分析还有很多,我们这里只涉及到SNV。同样的,突变的注释也涉及到多种软件,我们这里只涉及到了SnpEff。学海无涯啊~~~从培养起自主学习的思维和习惯开始。
3、拿到最终的注释文件(.vcf),我们还可以用到maftools、ComplexHeatmap等R包将突变情况进行可视化。
4、不急不慢,不慌不忙,坚持下去❤