ATAC-seq Analysis
ATAC-seq旧版处理流程
ATAC-seq Analysis
表观测序数据中比较重要的就是ATAC-seq、Chip-seq。这两者的原理和分析过程类似。
具体的异同详见此链接:https://www.jianshu.com/p/87bc2002e82c
(一)数据下载
数据下载是我走过的最曲折的路
强烈不推荐wget、curl——需要向上天再借500年才能等的起。
不推荐prefetch——不知道为什么prefetch在我这里也不起作用!按理说有ascp环境后,prefetch会自动调用ascp进行数据下载呀!可是prefetch依然显示通过https进行数据下载,这就没法下载呀!直接timeout!
正解——调用ascp下载数据
(1)ascp下载NCBI的SRA文件
此方法亲测不行,但是就在2个月前还是可以的。官网给出的解释大致意思就是——不好意思,这段时间出现无法通过ascp下载NCBI数据属于正常现象…blablabla…
(2)ascp下载EBI的fastq文件(强烈推荐,省去了SRA转成fastq的麻烦,很耗资源,很耗时)
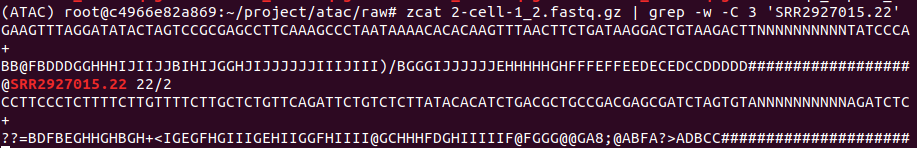
在此次数据分析中,下载SRR2927015、SRR2927016、SRR2927018、SRR3545580对应的fastq文件进行ATAC-seq数据分析的实战。
EBI官网输入对应SRR号(以SRR3545580为例),找到相应的fastq文件位置,复制连接地址。
http://ftp.sra.ebi.ac.uk/vol1/fastq/SRR354/000/SRR3545580/SRR3545580_1.fastq.gz
http://ftp.sra.ebi.ac.uk/vol1/fastq/SRR354/000/SRR3545580/SRR3545580_2.fastq.gz
下载方式如下(下载速递可达到每秒M级别,快的时候能到60M+,亲测此方法为目前最可取的方法)
1 | ascp -QT -l 300m -P33001 -i /home/gongyuqi/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR354/000/SRR3545580/SRR3545580_1.fastq.gz . |
最终下载结果
为了后续分析,将其改名为下
2-cell-1 SRR2927015
2-cell-2 SRR2927016
2-cell-4 SRR2927018
2-cell-5 SRR3545580
附上测序数据下载方式的相关参考资料
https://www.jianshu.com/p/df34b99cbd43
https://www.bioinfo-scrounger.com/archives/171/
(二)自定义docker容器的启动与数据共享
之前创建了具备ATAC-seq数据分析所需环境的docker容器,并push到阿里云。在此只需要pull该镜像,run即可使用。
但是!!!创建容器时并没有装ascp,因为只有普通用户才可以运行ascp。所以数据下载是在ubuntu主机进行的。为了能够在容器中使用下载的测试数据,运用数据卷,实现主机和容器间的数据共享。
1 | # 查看镜像 |

1 | # 启动容器实现数据共享 |
1 | # 查看容器内是否已经存在fastq数据 |

1 | # 激活ATAC环境(该环境配置有处理ATAC-seq数据的所需软件) |

(三)、数据过滤
运用软件 Trim Galore
去除接头和质量不合格的reads
Trim Galore是对FastQC和Cutadapt的包装。适用于所有高通量测序,包括RRBS(Reduced Representation Bisulfite-Seq ), Illumina、Nextera 和smallRNA测序平台的双端和单端数据。
Trimmomatic是针对Illumina高通量测序平台设计的接头去除和低质量reads清洗软件。
附上trim-galore相关参考资料: https://www.jianshu.com/p/7a3de6b8e503
以下列数据为例
2-cell-1 SRR2927015
1 | trim_galore -q 7 --phred33 --length 35 -e 0.1 --stringency 4 --paired -o /root/project/atac/clean/ 2-cell-1_1.fastq.gz 2-cell-1_2.fastq.gz |
trim_galore
–q:设置线程
–phred33:使用ASCII+33质量值作为Phred的分
–length:去除长度小于参数值的reads
-e:允许的最大误差
–stringency:设置与接头重叠的序列
–paired:对于双段测序文件,正反链质控通过才保留
-o:输出目录
结果如下

因为本次数据是用本机(8线程,16G)跑的,要给机子休息的时间,同时实验样本少,所以一个一个样本手动跑。
但是!当样本量多了,在服务器上跑的时候,循环就要省事很多,比如跑的时候去睡个觉…
拿本次样本写个循环,举个例
1 | ls /root/project/raw/*1.fq.gz >1 |
(四)、质量控制
测试数据的质控真的非常重要!!!如果测序质量本身就很好,没有质空这一步也影响不大。如果测序质量不好,不进行质控的化,直接影响后续的比对率。一般情况下,90%+的比对率才算比较正常,太低的比对率往往是质控步骤有问题,会非常严重的影响最终结果。亲测过!!!!
运用软件fastqc、multiqc
进行批处理
- 数据质控
1 | cd /root/project/atac/qc && mkdir rawqc trimqc |
(1)原始数据质控结果

(2)过滤后数据质量控制结果

- 质控结果整合
1 | # 原始数据qc结果整合 |
(1)原始数据整合结果

(2)过滤数据整合结果

附上multiqc相关参考资料
https://blog.csdn.net/ada0915/article/details/77201871
- mulitiqc整合结果解读
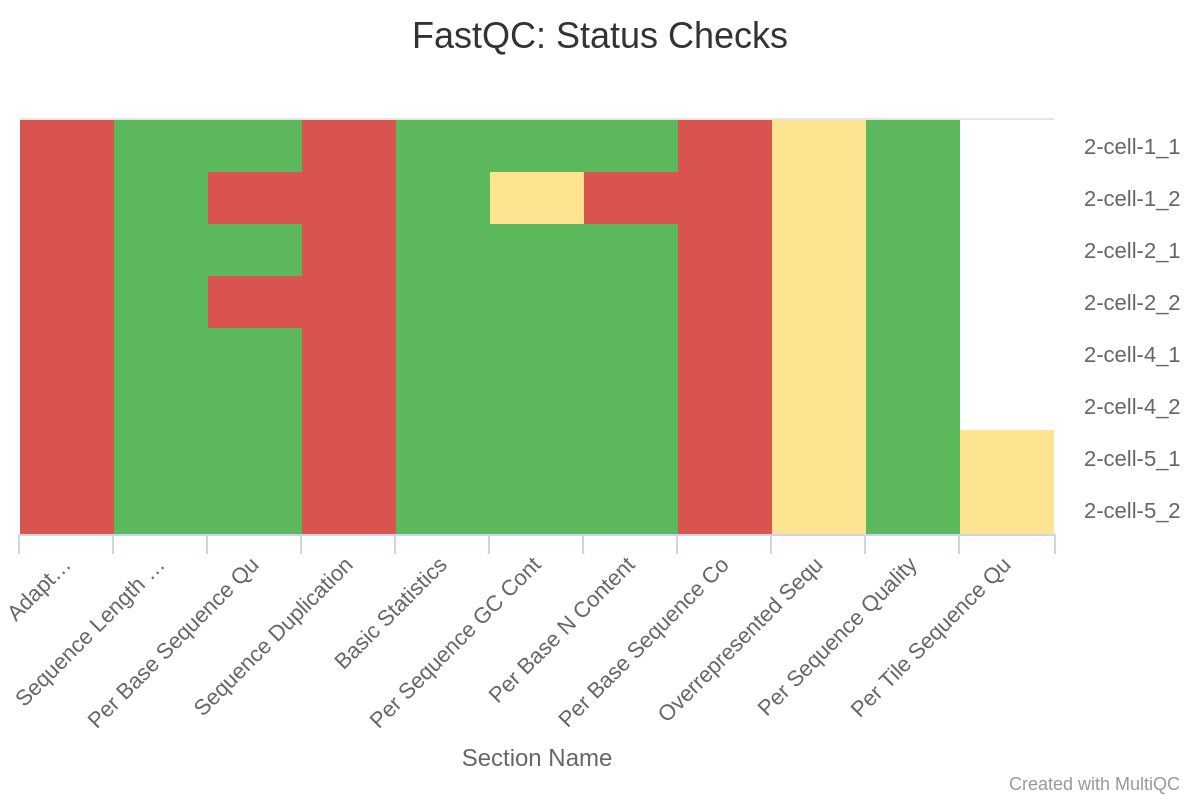
(1)过滤前
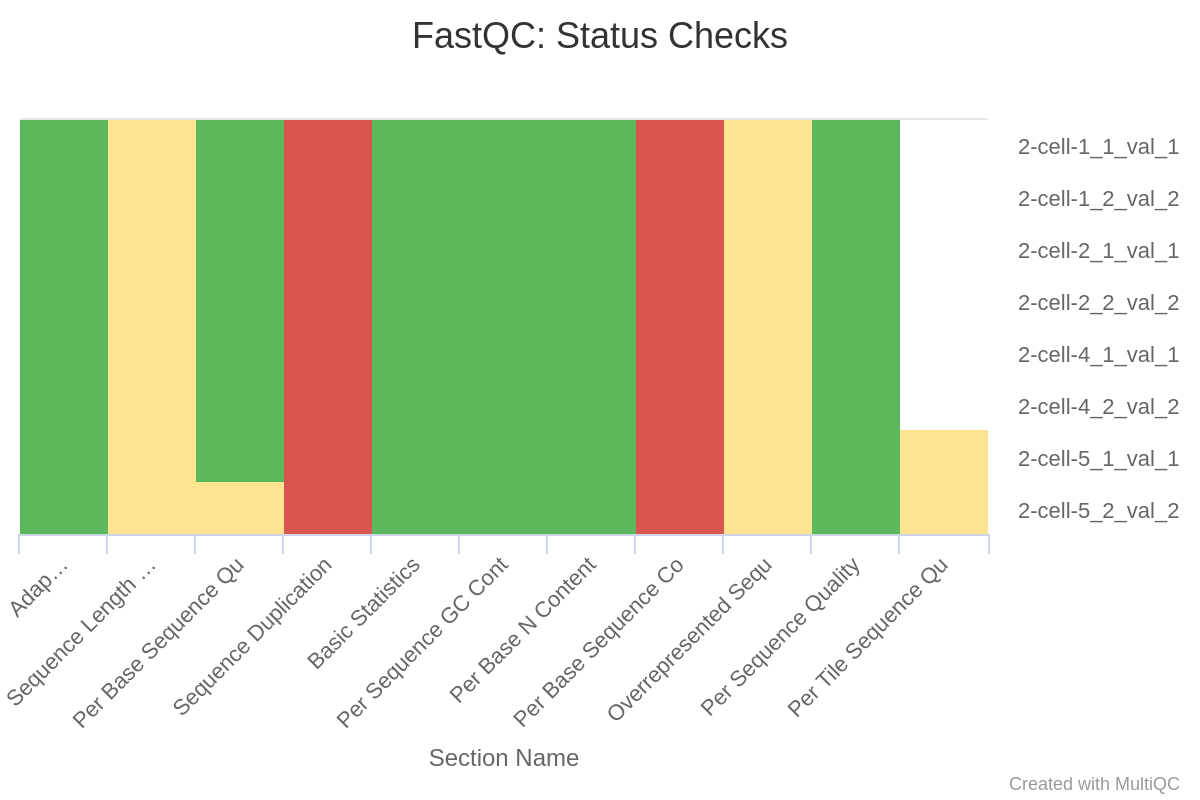
(2)过滤后
(3)解读(随便一搜,各种解读教程实在太多了~)
trim-galore完美的去除了adaptor;去除了质量不合格的序列使得2-cell-1_2和2-cell-2_2的质量过关;2-cell-1_2的N含量超标的序列被去除了。
因为去除了adptor和质量不合格的序列,所以sequence length自然是不一致的了。另外,还没有PCR去重,所以sequence duplication是不过关的,这一点会在PCR去重步骤中得到解决。至于Per Base Sequence Content,前10个碱基总是异常的波动,比对序列长度远大于10,处理时也可以设定不比对前10个,所以这个关系不大。
(4)以Sequence Length和Per Base N Content展示过滤前后的区别
过滤前
过滤后

(五)比对参考基因组
运用软件bowtie2、bedtools
一般对于DNA数据,比对软件一般使用:bowtie2、bwa
对于RNA数据,比对一般使用:hisat2、star
使用bedtools将最终的bam文件转成bed文件
(1)参考基因组及其注释文件的下载
参考基因组(解压后)
参考基因组注释文件(解压后)
本次分析用到的是小鼠的参考基因组及其注释文件
mm10.fa、gencode.vM24.annotation.gtf
(2)参考基因组索引构建(将构建的参考基因组索引文件cp到容器中的/root/project/atac/reference/mm10_index目录下)
1 | bowtie2-build /home/gongyuqi/biosoftware/reference/genome/mm10.fa mm10_index |
(3)参考基因组的比对,直接生成bam文件
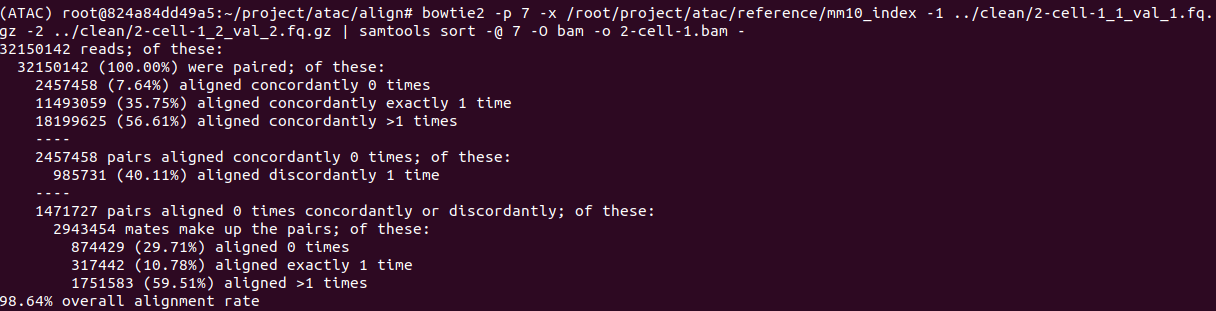
1 | bowtie2 -p 7 -x /root/project/atac/reference/mm10_index -1 ../clean/2-cell-1_1_val_1.fq.gz -2 ../clean/2-cell-1_2_val_2.fq.gz | samtools sort -@ 7 -O bam -o 2-cell-1.bam - |
bowtie2直接比对的结果会生成sam文件,sam文件很大,比较占空间。在比对这一步就直接利用samtools sort将其转成bam文件。
另外,samtool排序过程中并不会丢弃一部分reads,但是排序后的bam文件相比排序前的体积会更小。
比对相关参考资料
https://www.jianshu.com/p/6ed1bfbb7b72
比对结果
以2-cell-1_1_val为例进行结果展示
(4)构建原始bam文件的索引文件、状态文件
构建索引文件
1 | ls *.bam | while read id; do samtools index -@ 7 $id; done |
.bai文件即位生成的索引文件
构建状态文件
1 | ls *.bam | while read id; do samtools flagstat -@ 7 $id > $id.stat; done |
(5)去除PCR重复、构建去重后的索引文件、状态文件、
sambamba软件去重,会同时生成了去重后的bam和bai文件
1 | ls *.bam | while read id; do sambamba markdup -r -t 7 $id rmdup.$id; done |
构建状态文件
1 | ls rmdup*.bam | while read id; do samtools flagstat -@ 7 $id > $id.stat; done |
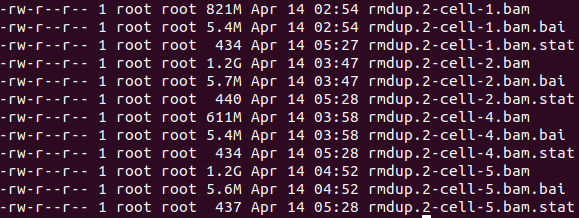
(6)去除同一样本的两条reads无法落在同一条染色体及测序质量低于30的reads、去除线粒体基因、构建索引文件、构建状态文件
去除不符合要求的reads及线粒体基因
1 | ls rm*.bam | while read id; do samtools view -@ 7 -h -f 2 -q 30 $id | grep -v chrM |samtools sort -O bam -@7 -o last.$id; done |
构建索引文件
1 | ls last*.bam | while read id; do samtools index -@ 7 $id; done |
构建状态文件
1 | ls last*.bam | while read id; do samtools flagstat -@ 7 $id > $id.stat; done |

(7)生成bed文件
1 | ls last*.bam | while read id; do bedtools bamtobed -i $id > $id.bed; done |

(六)使用macs2找peaks
使用软件macs2
以last.rmdup.2-cell-1.bam.bed进行演示
1 | macs2 callpeak -t last.rmdup.2-cell-1.bam.bed -g mm --nomodel --shift -100 --extsize 200 -n 2-cell-1 --outdir ../peaks/ |
最终生成文件
peaks注释
1 | setwd("path\\to\\.bed") |
(1)拿到每个样本中peaks对应得基因名
这一步非常重要,拿到基因名就可以根据课题需要进行差异分析等
1 | genelist<-peakAnno@anno@elementMetadata@listData[["SYMBOL"]] |
(2)单个文件作图
图片就不做展示了
1 | plotAnnoPie(peakAnno) |
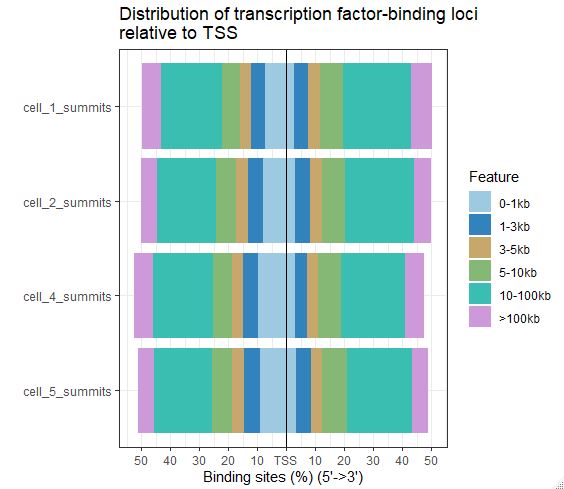
(3)plotAnnoBar和plotDistToTSS这两个柱状图都支持多个数据同时展示,方便比较
1 | setwd("path\\to\\.bed") |

1 | plotDistToTSS(peakAnnoList, title = "Distribution of transcription factor-binding loci\nrelative to TSS") |
(4)查看peaks在全基因组的位置(以2-cell-1样本为例)
1 | chropeaks<-readPeakFile("2-cell-1_peaks.narrowPeak") |

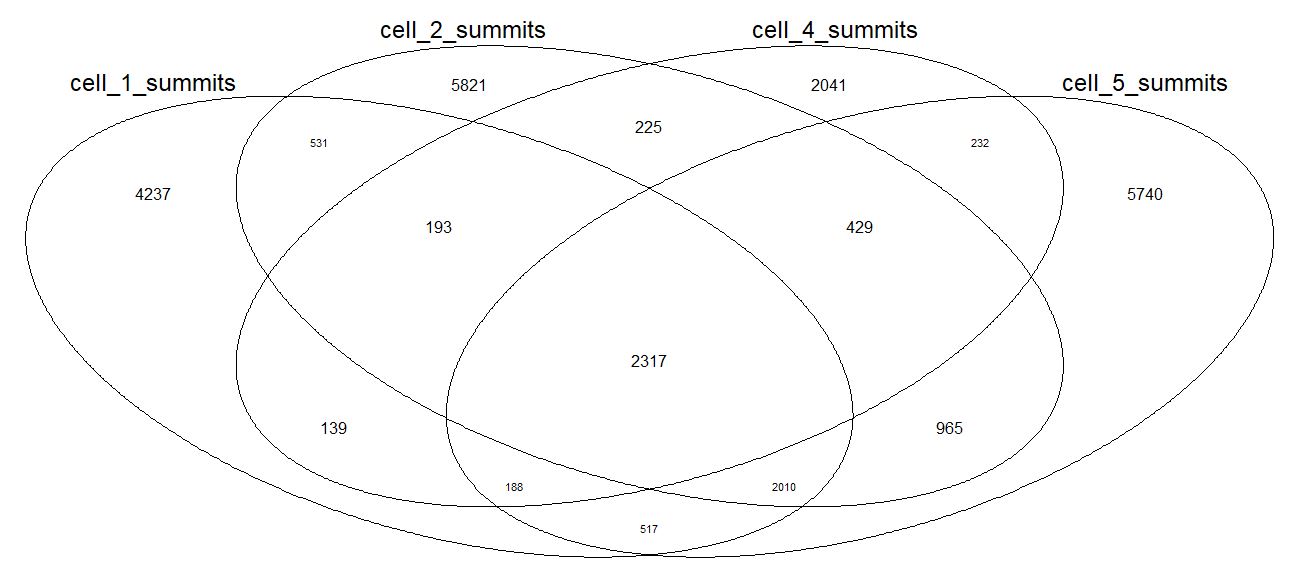
(4)重复样本间共同的peaks和注释基因
也可以用拿到的各个样本的基因名,做韦恩图,会比这个图更美观。
1 | genes<-lapply(peakAnnoList, function(i) as.data.frame(i)$geneId) |
peaks注释相关参考资料
ATAC-seq Analysis