Cellranger 软件流程
(一)环境搭建 下载Cell Ranger,下载10X GENOMICS配套的参考基因组.官网也提供了Cell Ranger Pipeline。
官网下载链接: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest
(1)下载、安装Cell Ranger软件 1 2 3 4 5 6 7 8 9 10 11 12 13 14 curl -o cellranger-6.0.1.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-6.0.1.tar.gz?Expires=1620565659&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cHM6Ly9jZi4xMHhnZW5vbWljcy5jb20vcmVsZWFzZXMvY2VsbC1leHAvY2VsbHJhbmdlci02LjAuMS50YXIuZ3oiLCJDb25kaXRpb24iOnsiRGF0ZUxlc3NUaGFuIjp7IkFXUzpFcG9jaFRpbWUiOjE2MjA1NjU2NTl9fX1dfQ__&Signature=IN508xcNVbAtoHul8SUog8KHmlAhg2lbR6-hCQeiwPs~-EfvTL1JmDurZ3dV-k3ry7TO9WP5Ae11yceDSzCfVIqglxHp6Pad4CftaStozIuEU7XJ0JbtDTO3DFVHRkpgNR8k48dNADA~JnhcHx8zOsT23VEeugqc7Z~35~05SOG8GJjxSJ8qGSTBdHYt7kPyqfZVs2apryxLOV-QBho66LvD4NLqNXFvzkPZ8t0e9Cv4rrV1JXseqY6PJnK8TPozTnDBnhfkop30FZ~jOLEY8wObOIA4b~EoyskSGjSfcvnp5urI7xjAyQNC6XtlJmRwDWEwWovgicScei2gkZqOGw__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA" tar zxvf cellranger-6.0.1.tar.gz echo 'export PATH=/home/gongyuqi/10X/cellranger-6.0.1:$PATH' >> ~/.bashrcsource ~/.bashrccellranger sitecheck > sitecheck.txt less -N sitecheck.txt cellranger testrun --id =testcellranger
标准输出最后两行为如下结果,表示cellranger可以正常使用
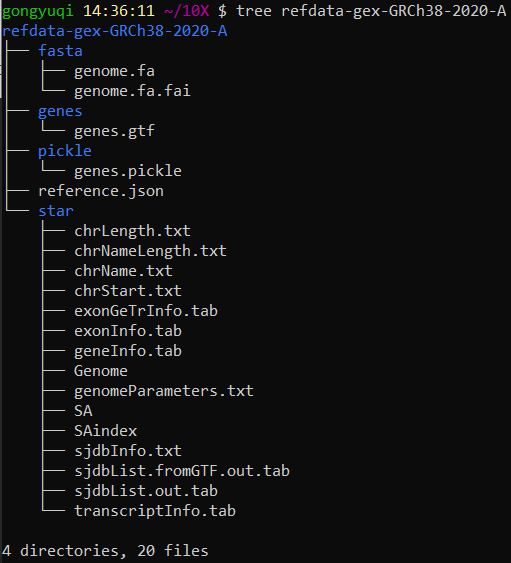
(2)下载人类参考基因组 1 2 curl -O https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz tar -zxvf refdata-gex-GRCh38-2020-A.tar.gz
查看一下文件的组成结构
(3)下载小鼠参考基因组 1 2 curl -O https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz tar -zxvf refdata-gex-mm10-2020-A.tar.gz
(二)下载测试数据 在这里要用到prefetch和ascp的组合,详细的软件安装以及测试数据下载请参考单细胞数据下载
(1)pretch下载SRA文件 1 2 3 4 5 cat sample.txt|while read id do nohup prefetch $id -O ./ && echo "${id} .sra done" &done
(2)ascp下载 1 2 3 ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/srr/SRR772/009/SRR7722939 ./ ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:vol1/srr/SRR772/002/SRR7722942 ./
(三)Cell Ranger 使用 (1)SRA格式转成fastq格式 1 2 3 4 5 cd /home/gongyuqi/project/scRNA-seq /raw_datals *.sra|while read id do nohup fastq-dump --gzip --split-files -A $id &done
(2)重命名文件 1 2 3 ls *.sra|while read id do mv ${id} _1*.gz ${id} _S1_L001_I1_001.fastq.gz; mv ${id} _2*.gz ${id} _S1_L001_R1_001.fastq.gz; mv ${id} _3*.gz ${id} _S1_L001_R2_001.fastq.gzdone
命名前
(3)质控 1 2 ls *R1*.gz > sample_fastq.txtls *R2*.gz >> sample_fastq.txt
(4)cellranger count cellranger是软件最核心的部分,可以完成细胞鉴定,基因组比对,过滤,UMI计数,细胞降维,聚类,差异分析等功能。内部流程很多,但是使用很简单。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 cd /home/gongyuqi/project/scRNA-seq /cellranger_countcat sample.txt|while read id ;do mkdir $id ;done cat sample.txt|while read id do cellranger count --id =$id \ --transcriptome=/home/gongyuqi/10X/refdata-gex-GRCh38-2020-A \ --fastqs=/home/gongyuqi/project/scRNA-seq /raw_data \ --sample=$id \ --nosecondary \ --localcores=16 \ --localmem=64 > ${id} .log 2>&1 done chmod +x cellranger_count.shnohup ./cellranger_count.sh &
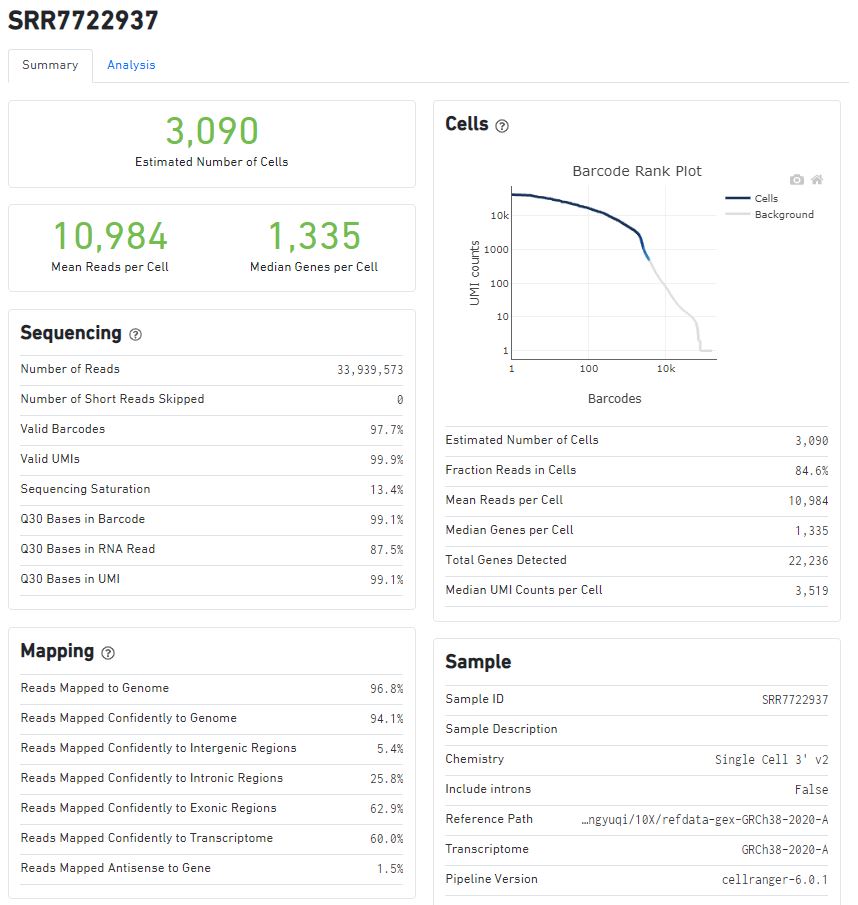
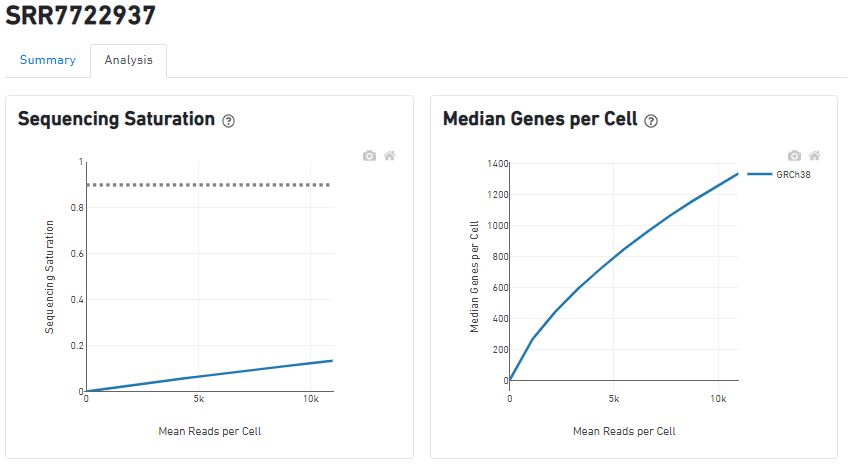
以SRR7722937为例,来看一下cellranger count的结果总结:Barcode,UMI,mapping情况,样本的信息(使用v2试剂,参考基因组,cellranger版本),reads与测序饱和度、基因数量的关系(正相关,情理之中~)
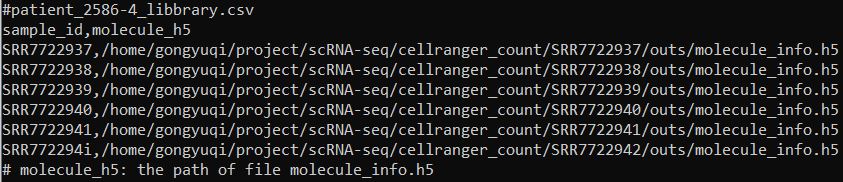
(5)cellranger aggr 第一步构建输入文件信息,文件内容如下。如果整合的样本既有v2试剂也有v3试剂,需要在第三列加上相应的试剂信息。如果都是用的同一种试剂,只需要保留前两列的信息即可。如本次测试数据。
1 2 3 4 5 cellranger aggr --id =patient_2586-4 \ --csv=patient_2586-4_libbrary.csv \ --normalize=mapped
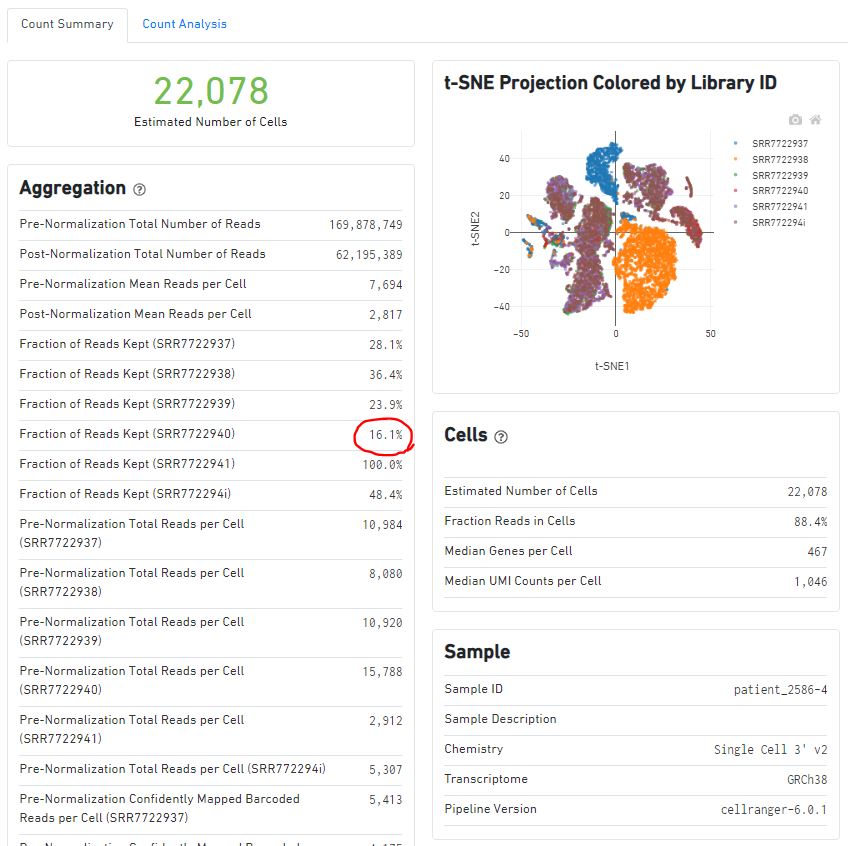
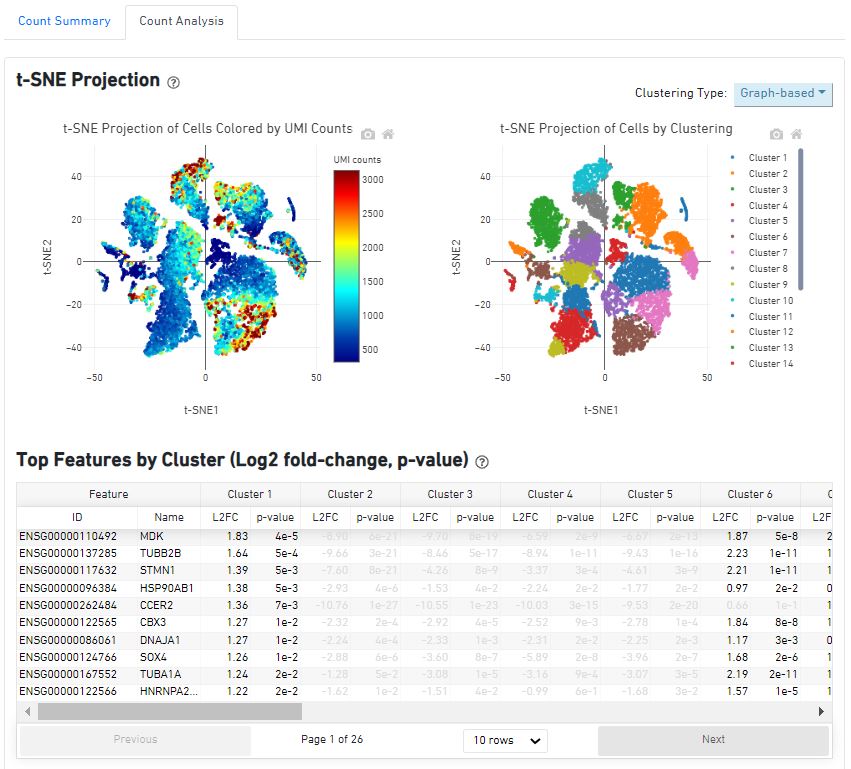
同样的,来查看一下cellranger aggr的结果报告。报告的消息解读可参考:https://www.jianshu.com/p/30de2aea4b74?utm_campaign=maleskine&utm_content=note&utm_medium=seo_notes&utm_source=recommendation
(四) Cell Ranger输出结果导入R cellranger aggar运行完成之后,在outs文件夹下面的子文件夹filtered_feature_bc_matrix中的三个文件上传到本地,解压导入R中。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 library( Matrix) barcode.path<- paste0( "barcodes.tsv" ) feature.path<- paste0( "features.tsv" ) matrix.path<- paste0( "matrix.mtx" ) mat<- readMM( file = matrix.path) feature.names<- read.delim( feature.path, header = FALSE , stringsAsFactors = FALSE ) barcode.names<- read.delim( barcode.path, header = FALSE , stringsAsFactors = FALSE ) colnames( mat) <- barcode.names$ V1 rownames( mat) <- feature.names$ V2 mat[ 1 : 4 , 1 : 4 ] dim ( mat)