可变剪切分析之MISO
RNA-seq高级分析之可变剪切
可变剪切分析之MISO
MISO软件原文档:https://miso.readthedocs.io/en/fastmiso/index.html#installing-fastmiso
数据下载
- 写一个bash脚本下载数据
一次性下载所有的.fastq.gz文件,本次实验数据是单端测序数据1
2
3
4
5
6#!/bin/bash
dir=/home/gongyuqi/.aspera/connect/etc/asperaweb_id_dsa.openssh
for id in {84,78,76,72,82,80,74,68}
do
nohup ascp -QT -l 300m -P33001 -i $dsa era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR974/SRR9749$id/SRR9749$id.fastq.gz . &
done - 执行脚本
1 | chmod +x download.sh |
比对
参考资料:http://www.360doc.com/content/18/0714/20/19913717_770401548.shtml
- tophat进行转录组的比对时,输出的是bam(二进制的sam)文件。
- tophat依赖bowtie2(bowtie1也可以),参考基因组索引用bowtie2-build进行构建。
- 如果FASTA格式的基因组与索引文件不在同一个文件夹下面,tophat会在运行中自动生成,所以记得把参考基因组和索引文件放在同一个文件夹下面,以减少系统运行时的高消耗。
- 如果GTF或者GFF格式的基因组注释文件存在, tophat会优先比对到注释文件的转录组上。最好事先准备好转录组索引以节约后续的比对时间。
- 如果没有事先准备好转录组索引,tophat会在运行过程中生成转录组索引,这样就比较耗时耗力了。
参考基因组索引构建、转录组索引构建
hg19参考基因组gtf文件下载链接:https://www.gencodegenes.org/human/release_36lift37.html
1 | #建立hg19参考基因组索引文件 |
参考基因组的比对
比对方法一
1 | #以其中一个样本为例,走一波比对转录组流程。 |
比对方法二
- 写一个比对的bash脚本文件(align.txt),如下
1
2
3
4
5
ls *.gz|while read id
do
nohup tophat -G ~/ref/hg19/gencode.v36lift37.annotation.gtf -p 30 -o ../aligndata/$id.out ~/ref/hg19/index/bowtie/hg19 $id &
done - 执行这个脚本,然后去睡觉,早上醒来看看比对得结果如何~运行结果
1
nohup bash align.txt &
每个文件夹打开结果如下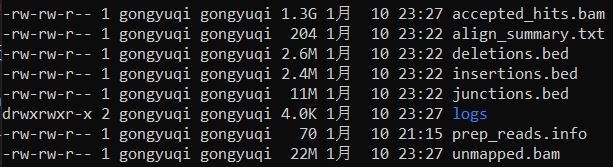
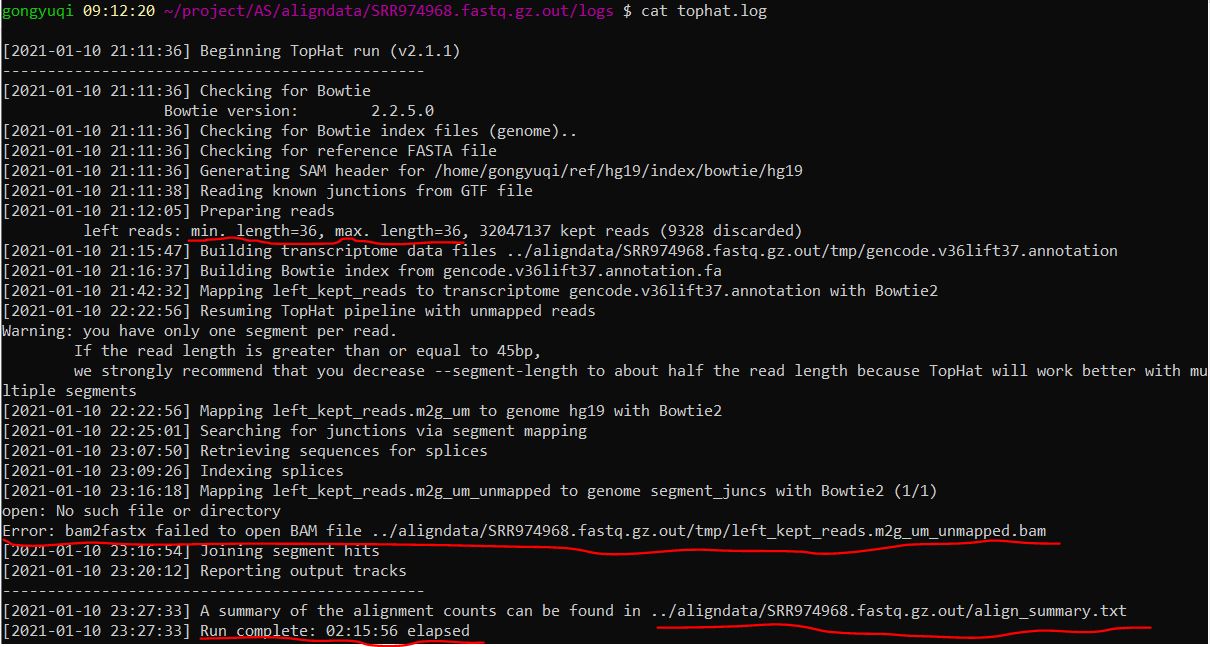
大概了解一下样本的运行日志
注意reads的长度——36。miso后续分析中需要知道这个值
注意其中有一个error,至于为什么有这个报错,先不追究
注意比对结果的那个统计文件align_summary.txt,查看这个文件发现各个样本比对情况都挺好的。这就是为什么这里暂时不追究那个报错
最后会统计样本比对时长
3、bam文件排序及索引文件建立
1 | ls *.bam|while read id; do samtools sort -@ 30 -m 8G -O bam $id -o ${id%%.*}.accepted_hits.sorted.bam;done |
运行MISO
下载hg19的GFF文件并构建索引
具体下载网页及详情:https://miso.readthedocs.io/en/fastmiso/annotation.html
1 | cd /home/gongyuqi/project/AS/annotation/human/hg19 |

这里我们以外显子的gff3为例构建索引
1 | index_gff --index SE.hg19.gff3 ./index |
结果如下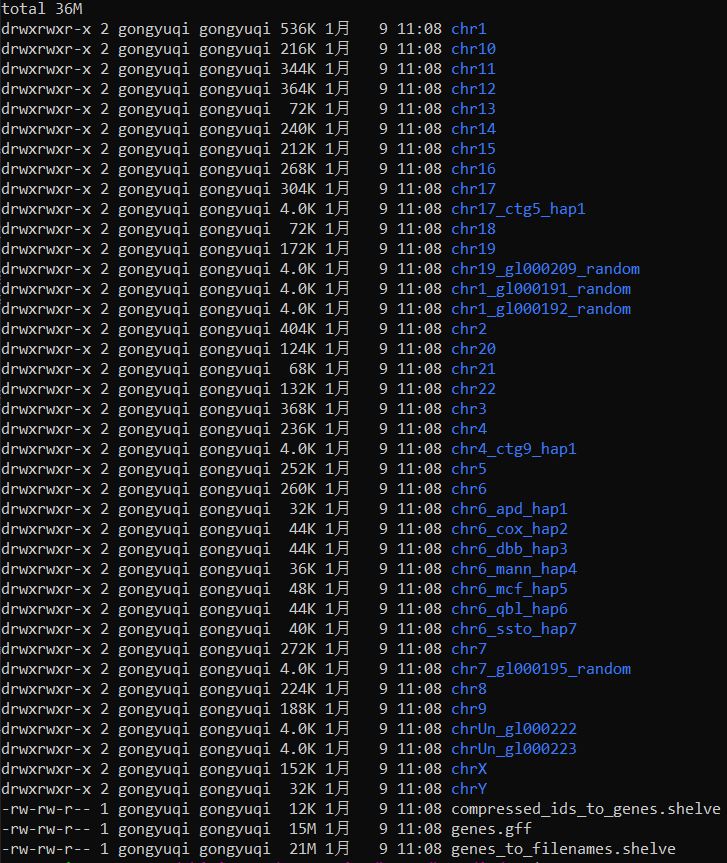
以单端测序样本为例运行miso
这一步生成的结果是很关键的,会计算alternative splicing events的PSI值。而这个在最后可视化的阶段会以posterior distributions over Ψ(PSI)的形式呈现——即sashimi_plot图最右边一列的柱状图。
写一个miso_run的脚本,内容如下
1
2
3
4
5
6
7
8
cd /home/gongyuqi/project/AS/aligndata/accpted_bam
index_db=/home/gongyuqi/project/AS/annotation/human/hg19/index/
out_dir=/home/gongyuqi/project/AS/miso_step1
ls *.sorted.bam|while read id
do
nohup miso --run $index_db $id --output-dir $out_dir/${id%%.*} --read-len 36 &
done运行脚本
1
nohup ./miso_run.sh &
举例查看其中一个样本的运行结果
| **样本间比较** |
这个软件最大的问题在于只能进行两个样本的比较,这一点就很气!!!
气归气,还是先运行一下,至少先把流程走完,先学起来。
1 | compare_miso --compare-samples SRR974978/ SRR974982/ comparison/ |
过滤
设置各种参数的阈值,目的是筛选差异显著的alternative splicing events。
1 | filter_events \ |
过滤前后.misoz_bf文件中的alternative splicing events有明显减少
可视化
1 | index_db=/home/gongyuqi/project/AS/annotation/human/hg19/index/ |
其中sashimi_plot_settings.txt是配置文件,内容如下。当然,也可以根据自己的作图需要修改相应的参数
1 | [data] |
可视化结果如下
因为我们选取的剪切事件是SRR974978和SRR974982之间的差异剪切事件,所以在多个样本间趋势不是很明显。但这里我们先学个方法。![]()
可变剪切分析之MISO