RNA-seq高级分析之可变剪切
可变剪切之rMART
官方参考资料
http://rnaseq-mats.sourceforge.net/
rMATS:https://github.com/Xinglab/rmats-turbo/blob/v4.1.0/README.md
rmats2sashimiplot:https://github.com/Xinglab/rmats2sashimiplot
(一)rMATS软件及相关依赖包的下载
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
conda create rMATs2.7 python=2.7
conda activate rMATs2.7
conda install -y Cython=0.29.14 numpy=1.16.5 blas lapack gsl=2.6 gcc=5.4.0 CMake=3.15.4
conda install libgfortran==1
deactivate rMATs2.7
cd /home/gongyuqi/project/AS/rMATS
wget https://github.com/Xinglab/rmats-turbo/releases/download/v4.1.0/rmats_turbo_v4_1_0_python_2_7.tar.gz
tar -zxvf rmats_turbo_v4_1_0_python_2_7.tar.gz
export PATH=/home/gongyuqi/project/AS/rMATS/rmats-turbo/:$PATH
source ~/.bashrc
|
(二)STAR比对软件的使用
这么多比对软件,为什么选STAR?
因为官网用的STAR!据说其他可以做可变剪切的比对软件生成的bam文件,rMATs运行不了~~~
1、构建参考基因组索引
1
2
3
4
5
6
|
conda activate alignment2.7
nohup STAR --runThreadN 30 --runMode genomeGenerate \
--genomeDir /home/gongyuqi/ref/hg38/index/star \
--genomeFastaFiles /home/gongyuqi/ref/hg38/Homo_sapiens.GRCh38.dna.primary_assembly.fa \
--sjdbGTFfile /home/gongyuqi/ref/hg38/Homo_sapiens.GRCh38.102.gtf &
|
2、用STAR软件进行比对
STAR的使用以及输出文件解读参考资料:
https://blog.csdn.net/yssxswl/article/details/105703869
https://www.jianshu.com/p/eca16bf2824e
1
2
3
4
5
6
7
8
9
10
11
12
|
cd /home/gongyuqi/project/AS/rawdata
ls *.gz|while read id
do
nohup STAR --runThreadN 20 \
--genomeDir /home/gongyuqi/ref/hg38/index/star \
--readFilesCommand gunzip -c \
--readFilesIn ./$id \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix /home/gongyuqi/project/AS/aligndata_star/${id%%.*}. &
done
|
3、比对情况统计并查看
(1)直接查看
1
| ls *.Log.progress.out|while read id;do sed -n '1,3p' $id;done
|
结果如下,比对情况还是不错的,可以进行后续的分析。
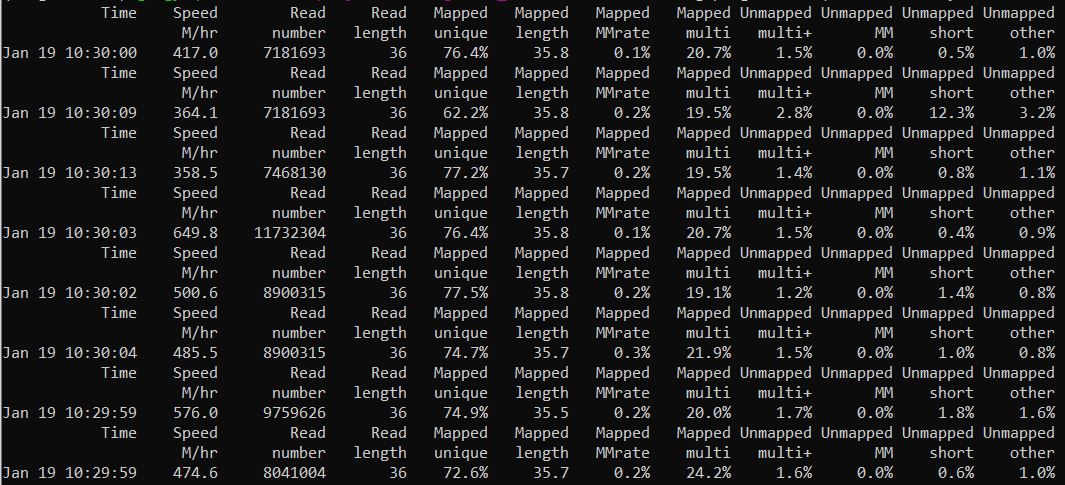
(2)flagstat统计比对情况查看比对情况
1
2
3
4
5
| ls *.bam|while read id
do
nohup samtools flagstat -@ 20 $id > ${id%%.*}.flagstat.txt &
done
ls *.txt|while read id;do cat $id | grep "mapped (";done
|
这个比对结果实在是难以置信!!但是结合上面的比对结果,还是可以相信比对这一步是没有问题的。
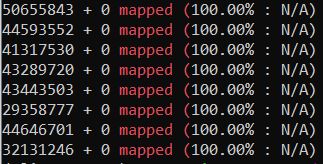
(三)运行rMATs
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
conda deactivate alignment2.7
conda activate rMATs2.7
cd /home/gongyuqi/project/AS/rMATS/rmats-turbo
input_dir=/home/gongyuqi/project/AS/aligndata_star
python rmats.py \
--b1 $input_dir/b1.txt \
--b2 $input_dir/b2.txt \
--gtf /home/gongyuqi/ref/hg38/ensembl/Homo_sapiens.GRCh38.102.gtf \
-t single --readLength 36 --nthread 20 \
--od /home/gongyuqi/project/AS/rMATS/rmats_output \
--tmp /home/gongyuqi/project/AS/rMATS/rmats_tmp
conda deactivate rMATs2.7
|
运行上面的代码,会报如下错误:

我查看了一下,/home/gongyuqi/miniconda3/envs/rMATs2.7/lib路径下有这个文件呢!为什么电脑找不到?触及到了我的知识盲区了~~~
查了一下午的资料,发现这个报错涉及到查找共享库(动态链接库)问题,根据查到的东西操作了一波居然没有解决,旁边的零食都不香了!😫
算了~~~我还是先去吃晚饭吧,已经饿得很瘦了!😫
。。。。。。
好啦,我吃完饭回来了。我继续倒腾,不放弃。
。。。。。。
额,我知道了,这段代码运行过程中还涉及到gcc编译,因为它依赖包中有GCC。
这两个问题实在是太专业了,今天又学习到了。现在有一点小小的开心!😊
解决方法参考资料:https://www.jianshu.com/p/a62e1d327023
1
2
3
4
5
|
export LD_LIBRARY_PATH=/home/gongyuqi/miniconda3/envs/rMATs2.7/lib
export LIBRARY_PATH=/home/gongyuqi/miniconda3/envs/rMATs2.7/lib
|
运行结果如下,关于这些结果文件的解释说明,可以参考rMATS官网,上面有很详细的阐述。
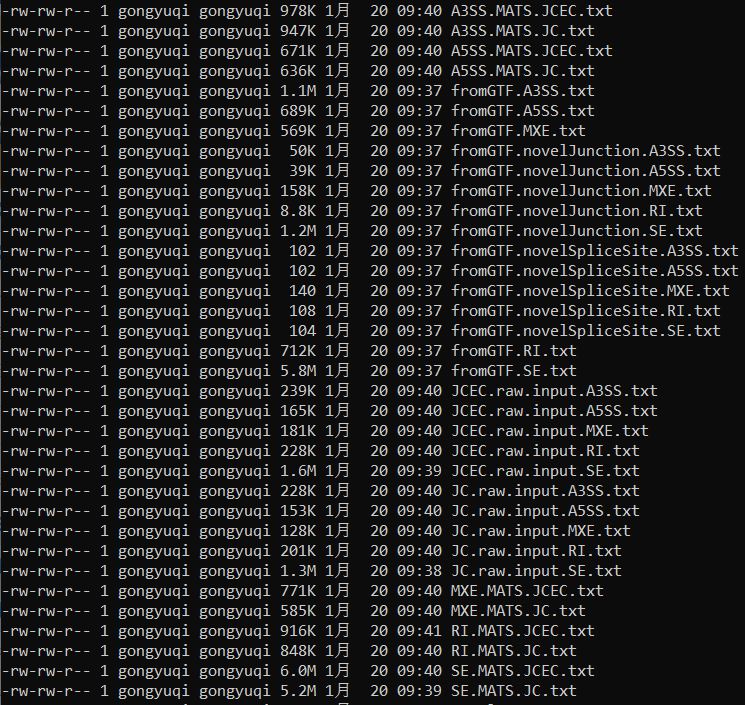
(四)rmats2sashimiplot可视化
终于到了激动人心的可视化阶段了,一顿操作之后,产生一张漂亮的图放PPT上还是很欣慰的。
1、环境搭建
1
2
3
| conda create -n rmats2sashimiplot python=2.7
conda activate rmats2sashimiplot
conda install -y numpy scipy matplotlib pysam samtools bedtools
|
2、准备可视化的txt文件
以Exon Skipping为例演示,这里我随便找一个基因进行可视化,注意了,这个基因不一定是差异可变剪切的基因哟,因为是我随便找的一个。嘻嘻🤭
1
2
| sed -n '2p' SE.MATS.JC.txt > SE.plot.PRMT2.txt
cat SE.plot.PRMT2.txt
|
文件内容如下:

3、可视化的txt文件
一开始,我非常自信的运行了下面的代码~
1
2
3
4
5
6
7
8
9
10
| gff3=/home/gongyuqi/ref/hg38/ensembl/Homo_sapiens.GRCh38.102.gff3
output_dir=/home/gongyuqi/project/AS/rMATS/rmats2sashimiplot/
rmats2sashimiplot \
--b1 SRR974968.Aligned.sortedByCoord.out.bam,SRR974972.Aligned.sortedByCoord.out.bam,SRR974978.Aligned.sortedByCoord.out.bam,SRR974984.Aligned.sortedByCoord.out.bam \
--b2 SRR974974.Aligned.sortedByCoord.out.bam,SRR974976.Aligned.sortedByCoord.out.bam,SRR974980.Aligned.sortedByCoord.out.bam,SRR974982.Aligned.sortedByCoord.out.bam \
-c chr21:+:46636438:46636547:$gff3 \
-e /home/gongyuqi/project/AS/rMATS/rmats_output/SE.plot.PRMT2.txt \
--l1 treatment --l2 control \
--exon_s 1 --intron_s 5 -t SE \
-o $output_dir/test
|
但是,我遇到了如下报错
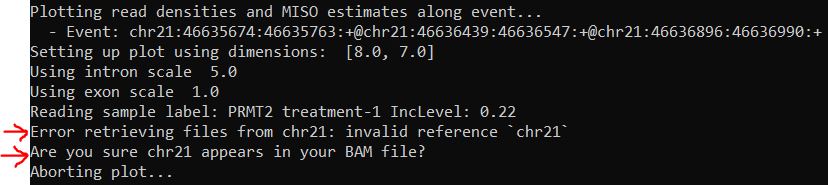
我回头看了一下我的bam文件,染色体是以1,2…X,Y形式体现而不是chr1,chr2…chrX,chrY形式体现。我第一次遇到染色体号的问题呢。我要怎么让bam文件中有chr存在且让其存在在正确的位置呢。
我绞尽脑汁的想了很久很久😫,我还考虑到是不是我参考基因组的问题,不同数据库参考基因组是不是有差异,我要不要换一个数据库的参考基因组。然后我一顿操作,无果~~~算了,我打一局游戏睡觉吧,明天再想。
起床吃早饭骑车去实验室思考查资料有一点小想法了我实践一下看看可不可
参考资料:https://www.jianshu.com/p/5ceda0350d0d
第一步:给bam文件添加chr
1
2
3
4
5
|
ls *.bam|while read id
do
nohup samtools view -h $id | sed -e '/^@SQ/s/SN\:/SN\:chr/' -e '/^[^@]/s/\t/\tchr/2'|awk -F ' ' '$7=($7=="=" || $7=="*"?$7:sprintf("chr%s",$7))' |tr " " "\t" | samtools view -h -b -@ 10 -S - > ${id%%.*}.Aligned.sortedByCoord.out.chr.bam &
done
|
第二步:使用添加chr的bam文件执行程序
1
2
3
4
5
6
7
8
9
10
| gff3=/home/gongyuqi/ref/hg38/ensembl/Homo_sapiens.GRCh38.102.gff3
output_dir=/home/gongyuqi/project/AS/rMATS/rmats2sashimiplot/
rmats2sashimiplot \
--b1 SRR974968.Aligned.sortedByCoord.out.chr.bam,SRR974972.Aligned.sortedByCoord.out.chr.bam,SRR974978.Aligned.sortedByCoord.out.chr.bam,SRR974984.Aligned.sortedByCoord.out.chr.bam \
--b2 SRR974974.Aligned.sortedByCoord.out.chr.bam,SRR974976.Aligned.sortedByCoord.out.chr.bam,SRR974980.Aligned.sortedByCoord.out.chr.bam,SRR974982.Aligned.sortedByCoord.out.chr.bam \
-c chr21:+:46636438:46636547:$gff3 \
-e /home/gongyuqi/project/AS/rMATS/rmats_output/SE.plot.PRMT2.txt \
--l1 treatment --l2 control \
--exon_s 1 --intron_s 5 -t SE \
-o $output_dir/SE.plot
|
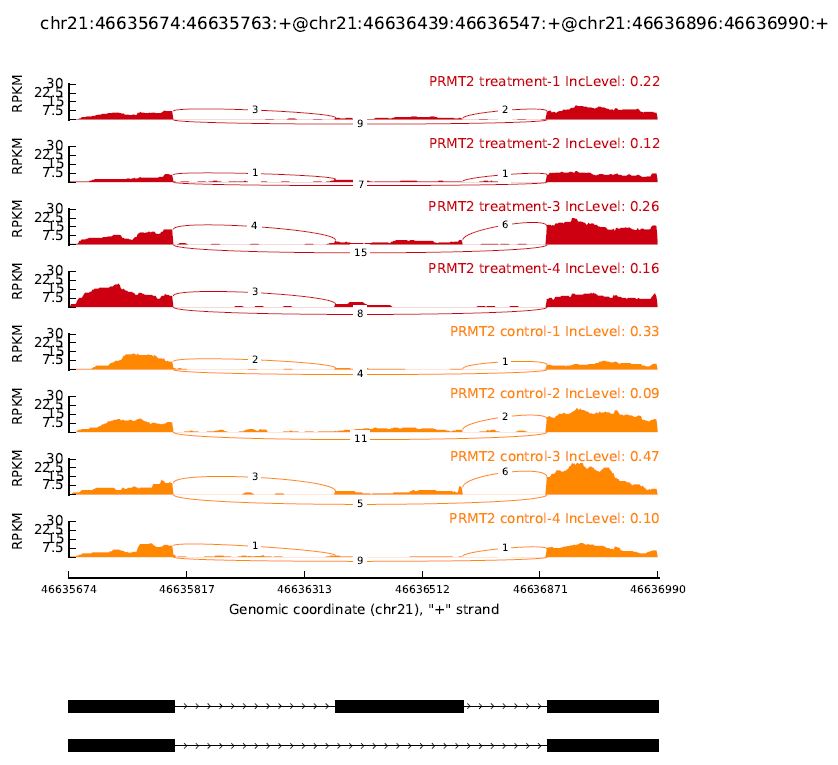
可视化结果如下(你会发现,咦~两组间差异不显著呀?因为我随便挑了一个基因可视化呀!我有偷偷可视化某个基因,可视化出来的差异性与我定量和半定量qPCR的结果一致哟,所以上述分析流程应该是OK的~~~嘻嘻嘻🤭不管怎样,这个基因确实存在Exon Skipping。)
总结
技术上
1、需要搭建两个重要的环境:运行rMATs的环境,运行rmats2sashimiplot的环境。
2、选用STAR进行参考基因组的比对(注意了,参考基因组和注释文件的版本要一致哟)。
3、在rMATs的环境下,结合上述生成的bam文件,运行rMATs软件,生成可变剪切事件的文件。
4、在rmats2sashimiplot环境下,可视化上述生成的可变剪切事件。
心态上
上午解决不了就下午解决,今天解决不了就明天解决。这个方法不行就换个方法。思考良久之后可以适当的问问老师。最关键的还是要按时吃饭按时睡觉。
SRR974972.Aligned.sortedByCoord.out.chr.bam,
1
2
3
4
5
6
7
8
9
10
| gff3=/home/gongyuqi/ref/hg38/ensembl/Homo_sapiens.GRCh38.102.gff3
output_dir=/home/gongyuqi/project/AS/rMATS/
rmats2sashimiplot \
--b1 SRR974968.Aligned.sortedByCoord.out.chr.bam,SRR974978.Aligned.sortedByCoord.out.chr.bam,SRR974984.Aligned.sortedByCoord.out.chr.bam \
--b2 SRR974974.Aligned.sortedByCoord.out.chr.bam,SRR974976.Aligned.sortedByCoord.out.chr.bam,SRR974980.Aligned.sortedByCoord.out.chr.bam,SRR974982.Aligned.sortedByCoord.out.chr.bam \
-c chr3:+:191375061:191375589:$gff3 \
-e /home/gongyuqi/project/AS/rMATS/ABC_GCB/rmats_output/SE.plot.CCDC50.txt \
--l1 ABC --l2 GCB \
--exon_s 1 --intron_s 5 -t SE \
-o $output_dir/CCDC50_SE_new.plot
|