scRNA-seq数据分析实战(三)
single cell RNA-seq && 10×
利用Seurate 2.3.3完成单细胞转录组下游分析,重现文章《Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA》的部分图片。
1 | rm(list = ls()) # clear the environment |
读入文章关于第一个病人的PBMC表达矩阵
1 | raw_dataPBMC <- read.csv('path/to/GSE117988_raw.expMatrix_PBMC.csv.gz', header = TRUE, row.names = 1) |

标准化
1 | dataPBMC <- log2(1 + sweep(raw_dataPBMC, 2, median(colSums(raw_dataPBMC))/colSums(raw_dataPBMC), '*')) # Normalization,矩阵大也会比较耗时 |
按照治疗阶段分组(后续会用到)
1 | timePoints <- sapply(colnames(dataPBMC), function(x) ExtractField(x, 2, '[.]')) |

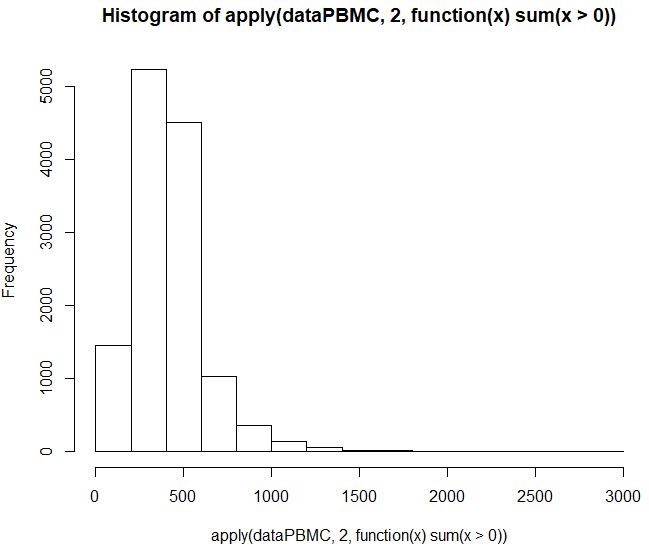
表达矩阵的基本信息可视化(可用于知道质控)
1 | ## 表达矩阵的质量控制 |
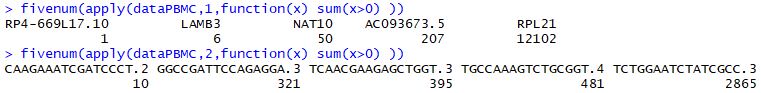

然后创建Seurat的对象
1 | # Create Seurat object |

根据需要添加metadata信息
1 | # Add meta.data (nUMI and timePoints) |
将对象进行基本的可视化
1 | sce=PBMC |

1 | GenePlot(object = sce, gene1 = "nUMI", gene2 = "nGene") |
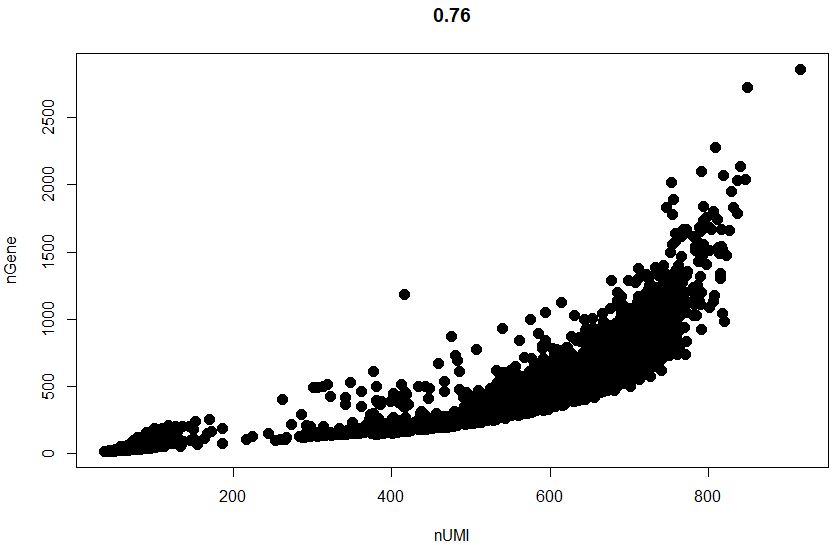
1 | #可以看看高表达量基因是哪些 |
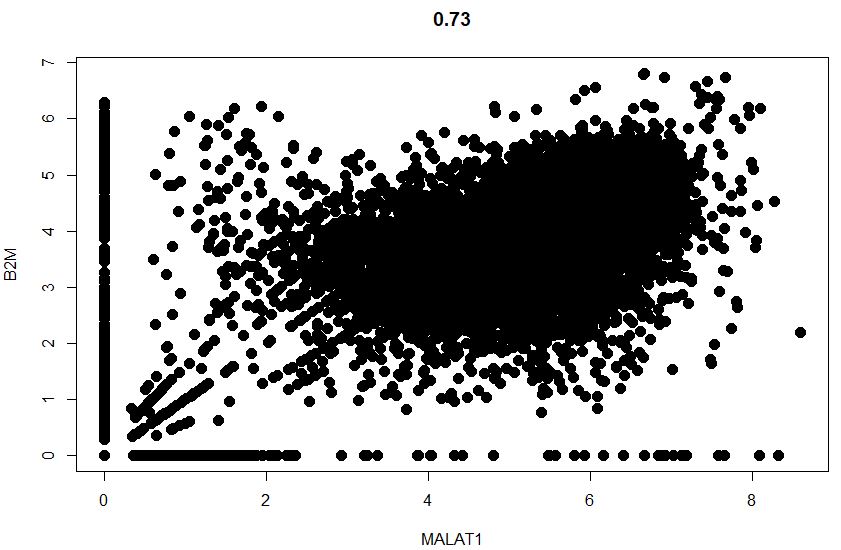
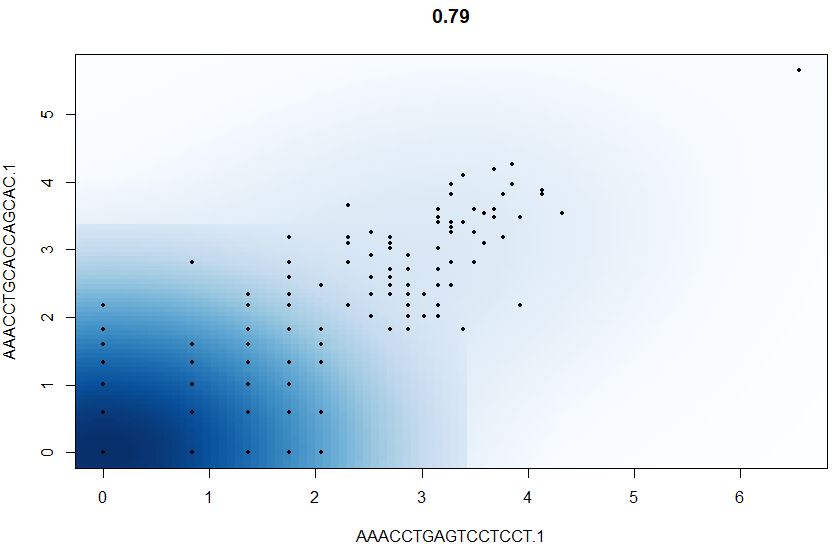
1 | # 散点图可视化任意两个细胞的一些属性(通常是基因的度量) |
聚类可视化(重点)
1 | # Cluster PBMC |
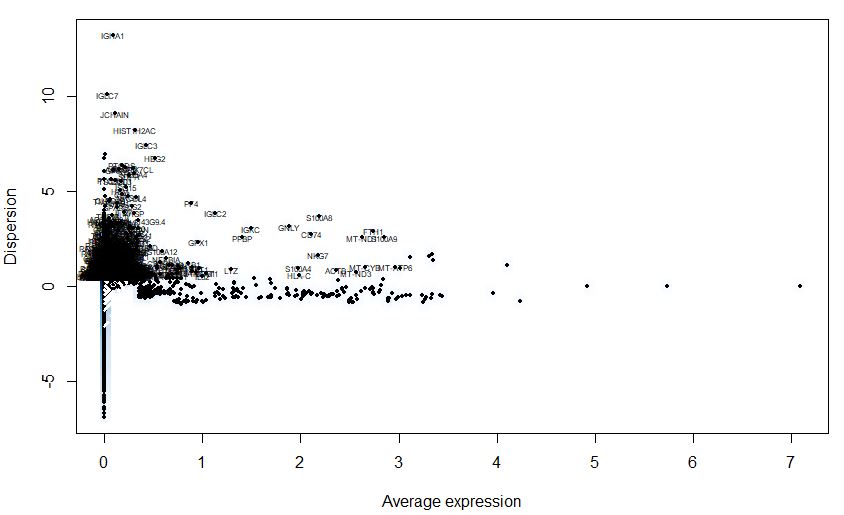
1 | PBMC <- RunPCA(object = PBMC, pc.genes = PBMC@var.genes) |
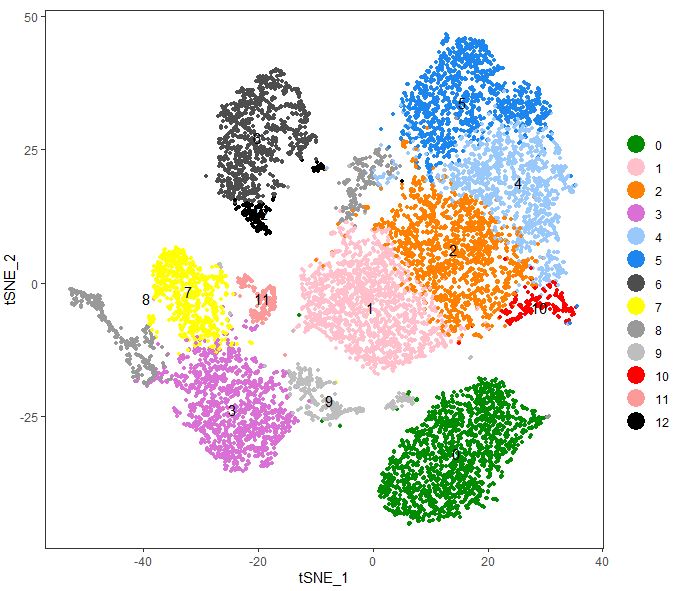
1 | table(PBMC@meta.data$TimePoints,PBMC@ident) |

添加亚群信息
1 | current.cluster.ids <- c(0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12) |
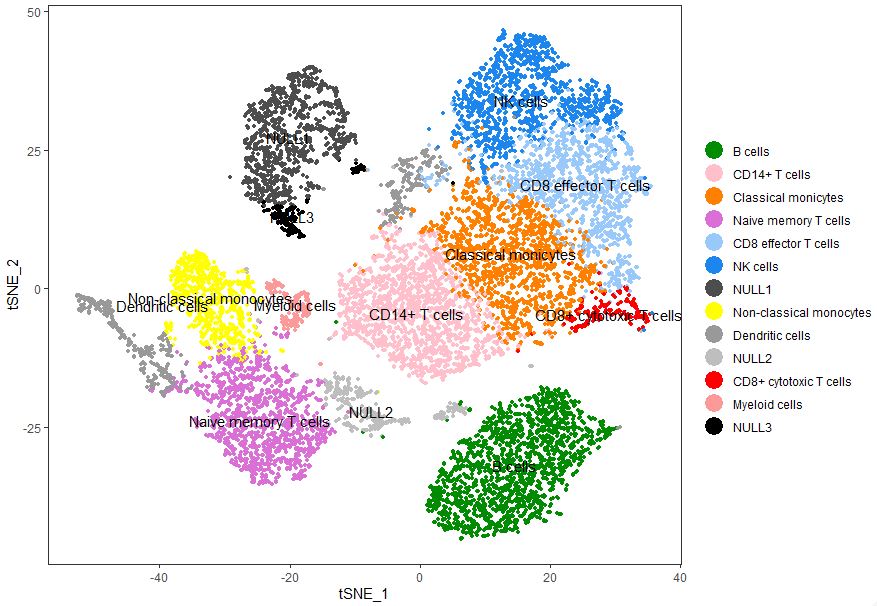
QUESTION
- 代码都跟原文的代码一样,虽然主要想体现的东西是有所体现的,聚类出来的细胞亚群还是有小小的偏差。
FindClusters的参数真的是我至今为止最最纠结的问题啦,参数不一样,亚群就会不一样,难不成为了得到自己想要的分群效果,不停的调整参数吗?- 对象问题也是个巨大的问题,对象里面包裹了好多东西,有些不太会调用。
- 该数据按照TimePoint来体现,目前自创的方法无法做到与原文类似的图,这也是一个待解决的问题。
scRNA-seq数据分析实战(三)