学习R包monocle对scRNA-seq数据进行分析
scRNA-seq数据分析(四)
创建数据集
后续分析的前提就是将数据构建成monocle需要的对象
因此这里先介绍一下monocle需要的用来构建CellDataSet对象的三个数据集
- 表达量矩阵
exprs:数据矩阵,行名是基因,列明是细胞编号
- 细胞的表型信息
phenoData:第一列是细胞编号,其他列是细胞的相关信息
- 基因注释
featureData:第一列是基因编号,其他列是基因对应的信息
这三个数据集要满足如下要求
表达量矩阵
- 保证它的列数等于
phenoData的行数
- 保证它的行数等于
featureData的行数
phenoData的行名需要和表达矩阵的列明匹配featureData和表达矩阵的行名要匹配featureData至少要有一列”gene_short_name”,就是基因的symbol
1
2
3
| rm(list = ls())
options(warn=-1)
suppressMessages(library(monocle))
|
这里同样使用scRNAseq R包中的数据集,构建monocle对象
1
2
3
4
5
6
7
|
library(scRNAseq)
data(fluidigm)
assay(fluidigm)<-assays(fluidigm)$rsem_counts
ct<-floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
sample_ann<-as.data.frame(colData(fluidigm))
|
1
2
3
4
5
6
7
|
gene_ann<-data.frame(
gene_short_name=row.names(ct),
row.names = row.names(ct)
)
pd<-new("AnnotatedDataFrame",data=sample_ann)
fd<-new("AnnotatedDataFrame",data=gene_ann)
|
#构建monocle后续分析的所有对象,主要是根据包的说明书,仔细探索其需要的构建对象的必备元素
#因为表达矩阵是counts值,所以注意expressionFamily参数,其他类型的值用其他参数
1
2
3
4
5
6
7
8
| sc_cds<-newCellDataSet(
ct,
phenoData = pd,
featureData = fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit = 1
)
sc_cds
|
在此示范如何加载加载RPKM数据,在此并不运行该部分代码
从本地读入RPKM值文件,构造CellDataSet对象
1
2
3
4
5
6
7
8
9
|
expression_matrix<-read.table("fpkm_matrix.txt")
sample_sheet<-read.delin("cell_sample_sheet.txt")
gene_annotation<-read.delin("gene_annotation.txt")
pd<-new("AnnotatedDataFrame",data=sample_sheet)
fd<-new("AnnotatedDataFrame",data=gene_annotation)
sc_cds<-newCellDataSet(as.matrix(expression_matrix),
phenoData=pd,featureData=fd)
|
monocle和scater、seurat他们基于的对象不一样,所以monocle还提供了转换函数
1
2
3
4
5
| lymphomadata<-sc_cds
lymphomadata_seurat<-exportCDS(sc_cds,'Seurat')
lymphomadata_scater<-exportCDS(sc_cds,'Scater')
|
下面是monocle对新构建的CellDataSet对象的标准操作
1
2
3
4
5
6
|
library(dplyr)
colnames(phenoData(sc_cds)@data)
sc_cds<-estimateSizeFactors(sc_cds)
sc_cds<-estimateDispersions(sc_cds)
|
质控
对基因和细胞进行质量控制,指控指标根据课题进行具体探索,没有统一标准,这里只是简单演示
过滤基因
1
2
3
4
5
6
7
8
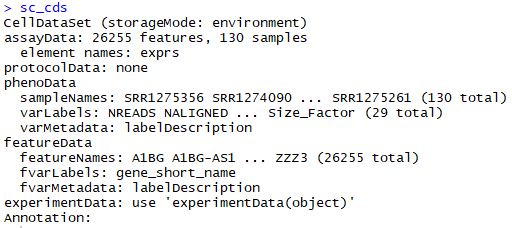
| cds<-sc_cds
cds
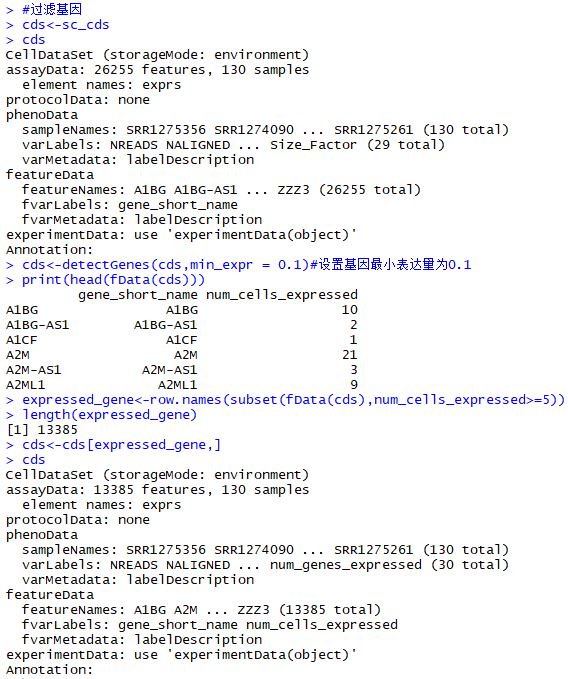
cds<-detectGenes(cds,min_expr = 0.1)
print(head(fData(cds)))
expressed_gene<-row.names(subset(fData(cds),num_cells_expressed>=5))
length(expressed_gene)
cds<-cds[expressed_gene,]
cds
|
过滤细胞
1
2
3
4
5
6
7
8
9
|
print(head(pData(cds)))
tmp<-pData(cds)
fivenum(tmp[,1])
fivenum(tmp[,30])
valid_cell<-row.names(pData(cds))
cds<-cds[,valid_cell]
cds
|

聚类
单细胞转录组最重要的就是把细胞分群,相关的算法非常多,这里选用最常用的tSNE
1
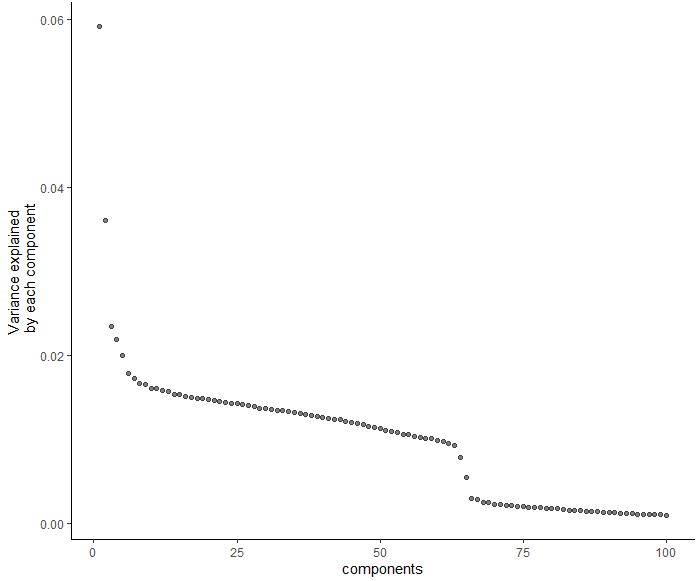
| plot_pc_variance_explained(cds,return_all = F)
|
1
2
3
4
5
6
|
cds<-reduceDimension(cds,max_components = 2,num_dim=6,
reduction_method = 'tSNE',verbose = T)
cds<-clusterCells(cds,num_clusters = 4)
plot_cell_clusters(cds,1,2,color="Biological_Condition")
table(pData(cds)$Biological_Condition)
|
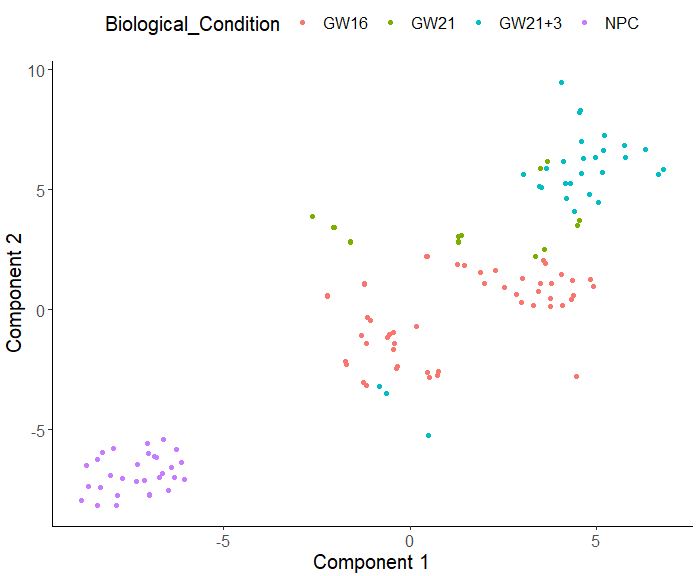

可以看出,用monocle的方法进行单细胞转录组数据分析,GW的三群细胞依然无法很好的区分开,NPC倒是聚类得非常完美
寻找差异基因
1
2
3
4
|
Sys.time()
diff_test_res<-differentialGeneTest(cds,fullModelFormulaStr = "~Biological_Condition")
Sys.time()
|
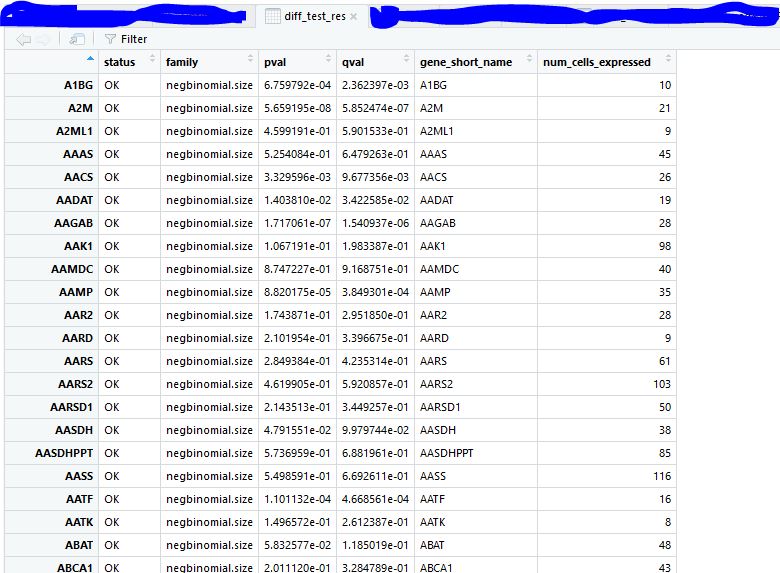
选出差异基因
1
2
| sig_gene<-subset(diff_test_res,qval<0.1)
head(sig_gene[,c("gene_short_name","pval","qval")])
|

1
2
3
4
5
6
7
| choosed_gene<-as.character(head(sig_gene$gene_short_name))
plot_genes_jitter(cds[choosed_gene,],grouping = "Biological_Condition",ncol = 2)
plot_genes_jitter(cds[choosed_gene,],grouping = "Biological_Condition",
color_by = "Biological_Condition",nrow = 3,ncol = NULL
)
|
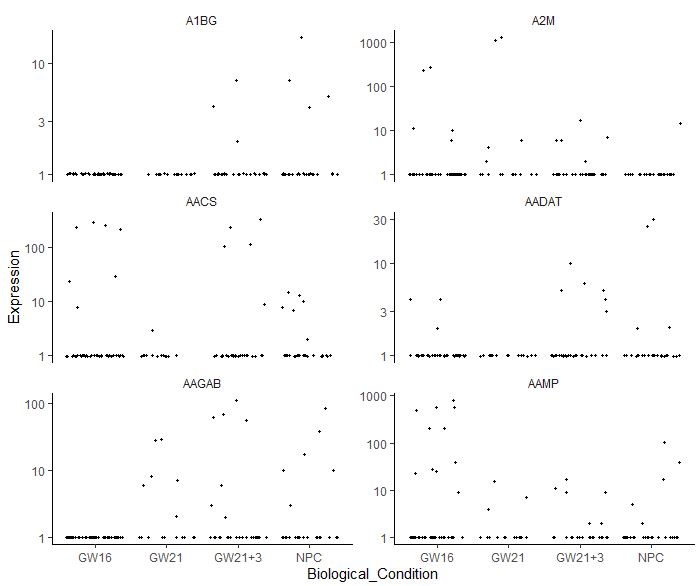
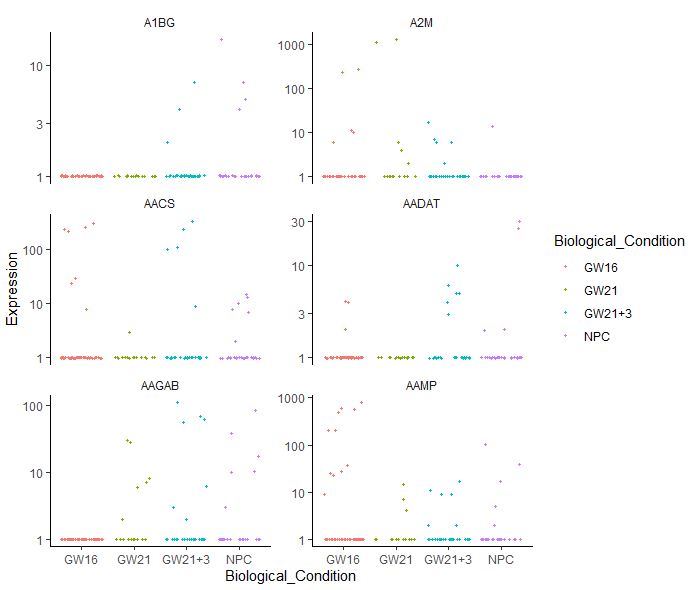
推断发育轨迹 这是monocle包最大的亮点
第一步:挑选合适的基因
有多种方法,例如可以提供已经的基因集,这里选取统计学上有差异的基因
1
2
3
| ordering_gene<-row.names(subset(diff_test_res,qval< 0.01))
cds<-setOrderingFilter(cds,ordering_gene)
plot_ordering_genes(cds)
|
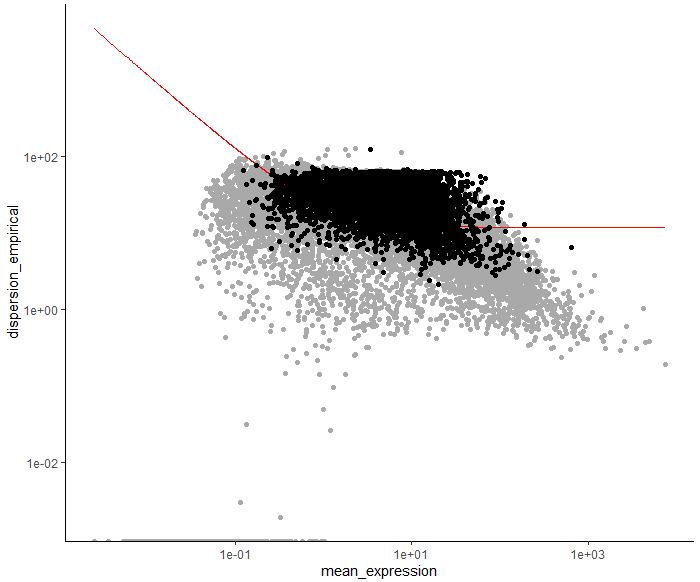
第二部:降维
降维的目的是更好的展示数据。函数里提供很多方法,不同方法最后展示的图会有所不同,DDRTree是Monocle2使用的默认方法
1
| cds<-reduceDimension(cds,max_components = 2,method='DDRTree')
|
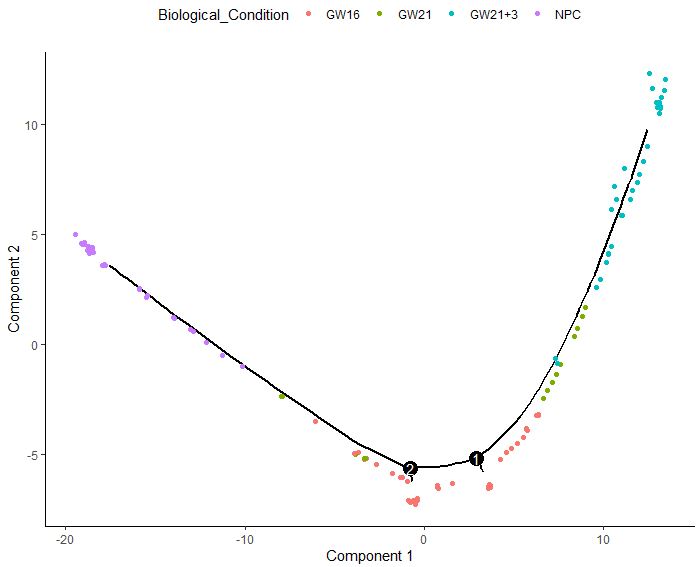
第三步:对细胞进行排序
1
2
| cds<-orderCells(cds)
plot_cell_trajectory(cds,color_by = "Biological_Condition")
|
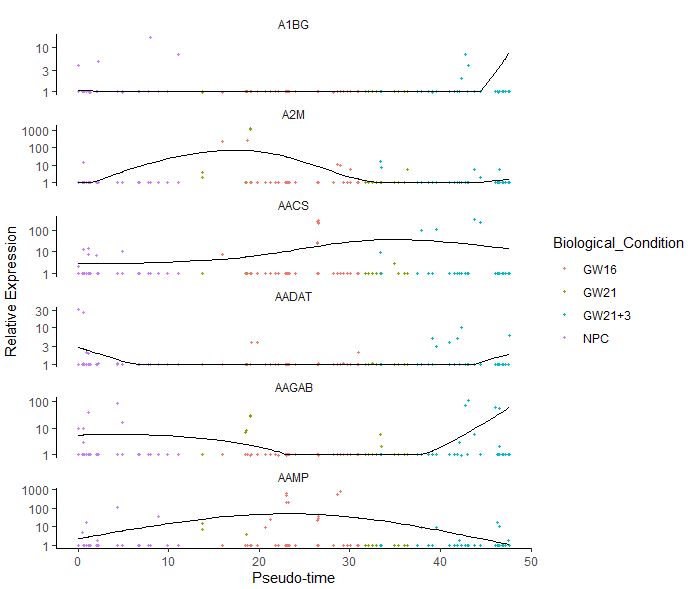
第四步:可视化marker gene随发育阶段变化的表达情况
1
2
|
plot_genes_in_pseudotime(cds[choosed_gene,],color_by = "Biological_Condition")
|
参考资料
生信技能数(B站、公众号)