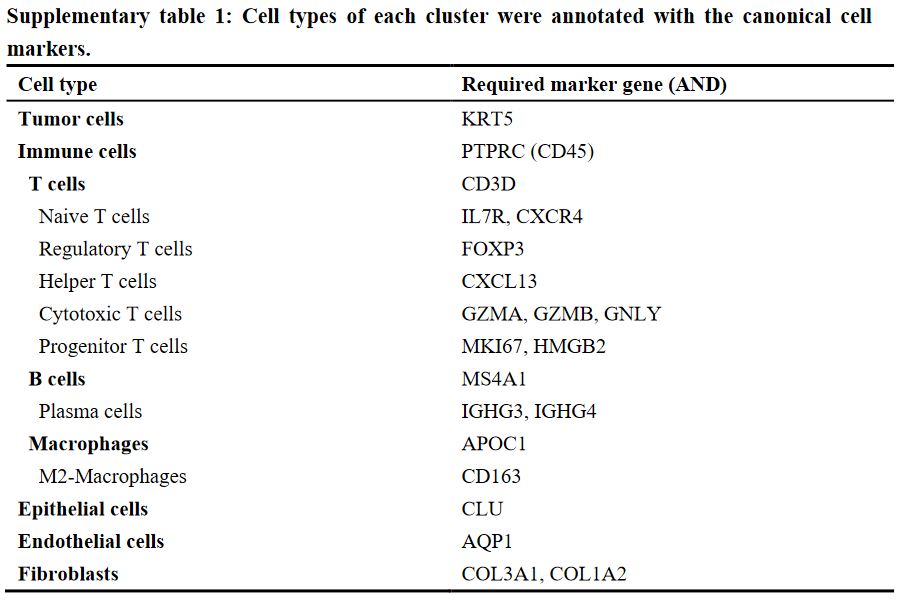
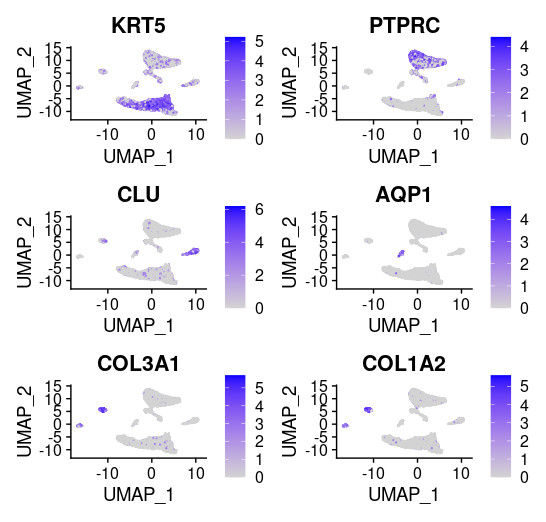
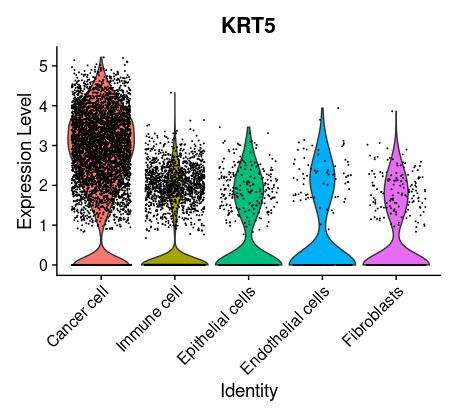
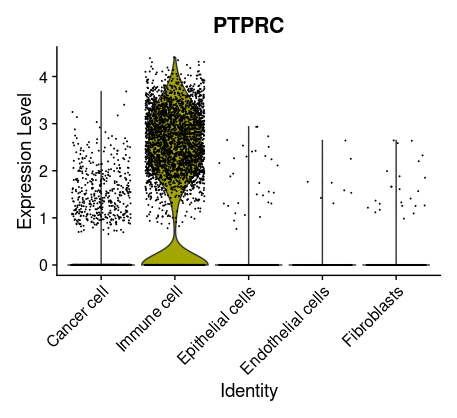
## =============== 去除线粒体基因 # The [[ operator can add columns to object metadata. This is a great place to stash QC stats #此次数据检测到大量线粒体基因 grep(pattern ="^MT\\.",rownames(tissu1),value =T) tissu1[["percent.mt"]]<- PercentageFeatureSet(tissu1, pattern ="^MT\\.") head(tissu1@meta.data,5) summary(tissu1@meta.data$nCount_RNA)
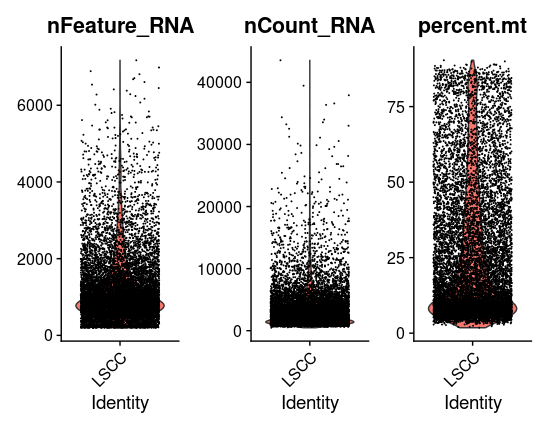
看一看过滤前数据情况
1 2 3 4 5 6 7 8 9 10 11 12 13
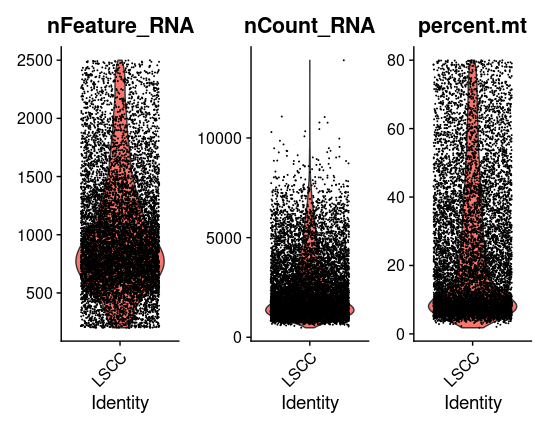
# Visualize QC metrics as a violin plot VlnPlot(tissu1, features =c("nFeature_RNA","nCount_RNA","percent.mt"), ncol =3) # density data<-tissu1@meta.data library(ggplot2) p<-ggplot(data = data,aes(x=nFeature_RNA))+geom_density() p
# FeatureScatter is typically used to visualize feature-feature relationships, but can be used # for anything calculated by the object, i.e. columns in object metadata, PC scores etc. plot1 <- FeatureScatter(tissu1, feature1 ="nCount_RNA", feature2 ="percent.mt") plot2 <- FeatureScatter(tissu1, feature1 ="nCount_RNA", feature2 ="nFeature_RNA") plot1 + plot2
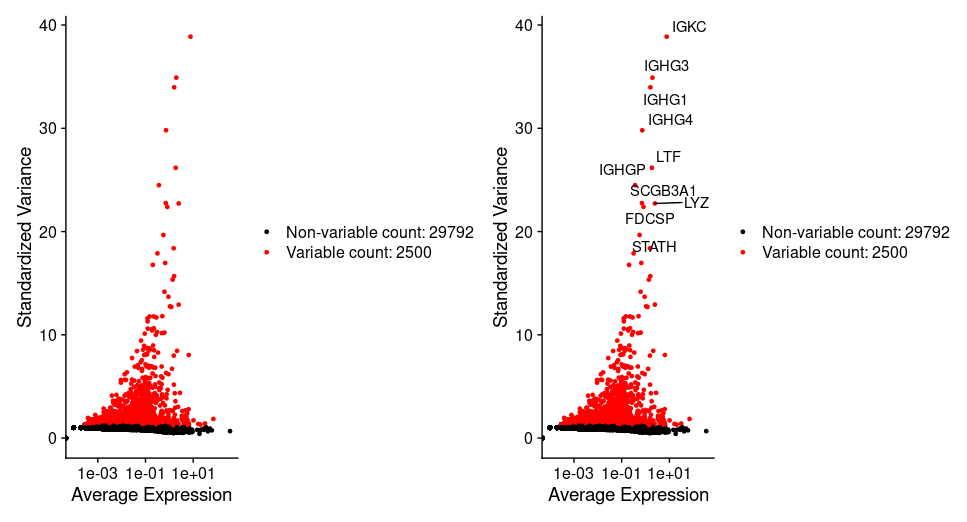
## =============== 1. Scaling the data #Seurat默认是做前2000个基因的归一化 #如何有热图展示需求的小伙伴,可以做全部基因的归一化,方便后续分析,我们这里就全部基因进行归一化 all.genes <- rownames(tissu1) tissu1 <- ScaleData(tissu1, features = all.genes)
## =============== 2. PCA降维 tissu1 <- RunPCA(tissu1, features = VariableFeatures(object = tissu1)) # Examine and visualize PCA results a few different ways print(tissu1[["pca"]], dims =1:5, nfeatures =5) #visualization VizDimLoadings(tissu1, dims =1:2, reduction ="pca") DimPlot(tissu1, reduction ="pca") DimHeatmap(tissu1, dims =1, cells =500, balanced =TRUE) DimHeatmap(tissu1, dims =1:20, cells =500, balanced =TRUE)
## =============== 3. PC个数的确定 #这个非常非常具有挑战性,针对这个问题有很多教程,我们这里不着重讨论,因为我们主要是图表复现 # NOTE: This process can take a long time for big datasets, comment out for expediency. More # approximate techniques such as those implemented in ElbowPlot() can be used to reduce # computation time tissu1 <- JackStraw(tissu1, num.replicate =100) tissu1 <- ScoreJackStraw(tissu1, dims =1:20) JackStrawPlot(tissu1, dims =1:20) ElbowPlot(tissu1)
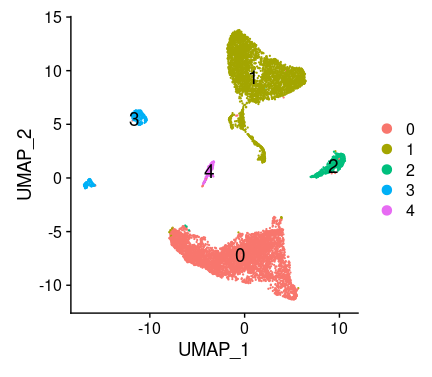
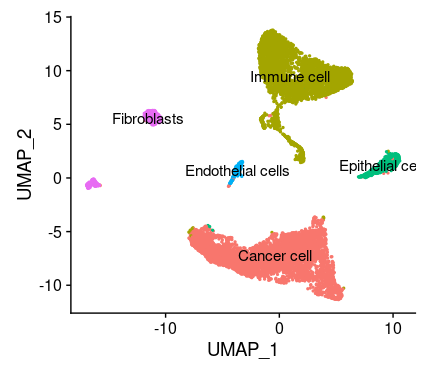
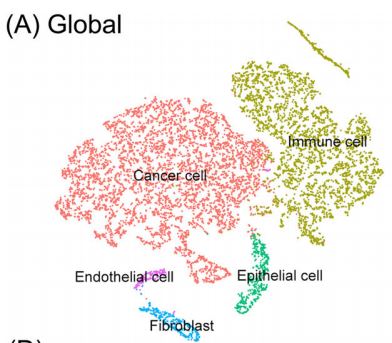
# If you haven't installed UMAP, you can do so via reticulate::py_install(packages = # 'umap-learn') tissu1 <- RunUMAP(tissu1, dims =1:10) # note that you can set `label = TRUE` or use the LabelClusters function to help label # individual clusters DimPlot(tissu1, reduction ="umap",label =T,label.size =5) #也可以tsne的方式展示细胞分区,但是umap给出的空间分布信息是有意义的,所以更倾向与使用umap #tissu1 <- RunTSNE(tissu1, dims = 1:10) #DimPlot(tissu1, reduction = "tsne",label = T,label.size = 5)
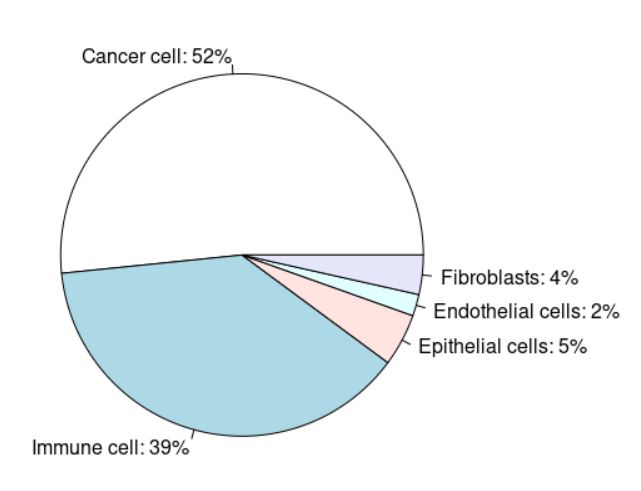
聚类结果,一共5个群
Step 7、差异表达
1 2 3 4 5 6
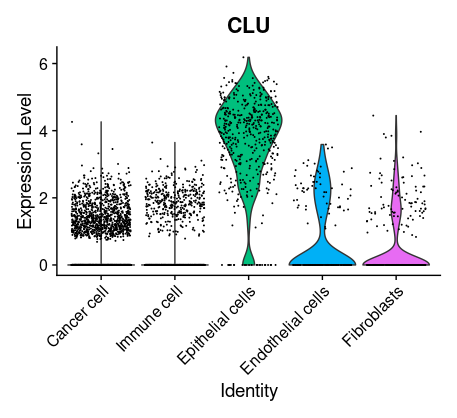
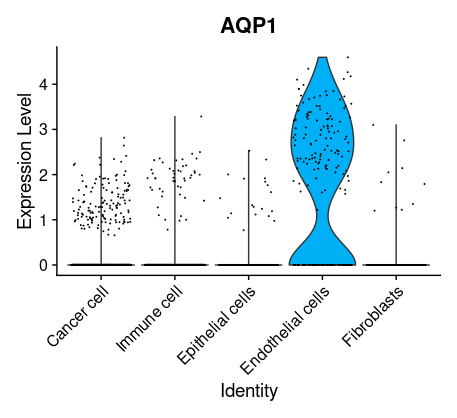
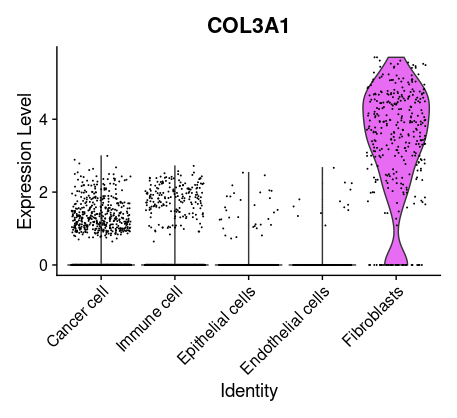
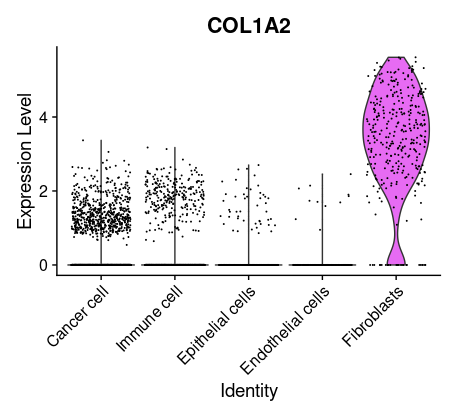
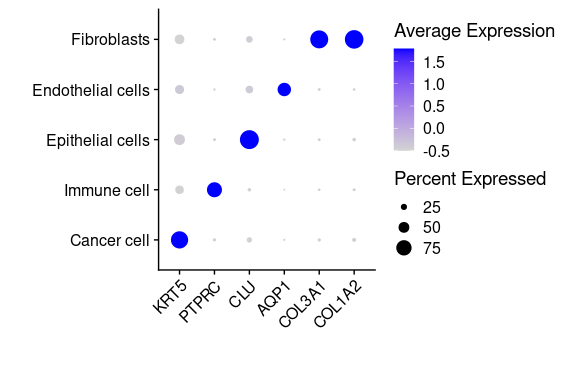
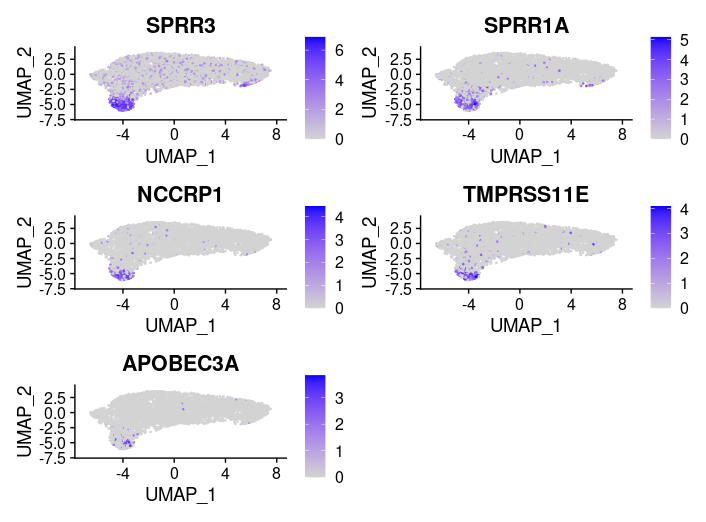
## =============== 差异表达及细胞类型鉴定 # find markers for every cluster compared to all remaining cells, report only the positive ones tissu1.markers <- FindAllMarkers(tissu1, only.pos =TRUE, min.pct =0.25, logfc.threshold =0.25) write.csv(tissu1.markers,file ="tissu1.markers.csv") #展示每一个cluster中top2显著的marker tissu1.markers %>% group_by(cluster)%>% top_n(n =2, wt = avg_log2FC)